多肽及蛋白质的人工合成(卷名:生物学)
chemical synthesis of peptides and protein
以氨基酸为原料,用化学方法合成多肽或蛋白质。其目的是:①确证天然多肽或蛋白质的结构;②生产天然的、在生物体内含量极微但有医疗或其他生物效用的多肽;③改变部分结构,研究其结构与功能的关系,并设计更有效的药物。
肽合成的基本原理 将两个氨基酸合成一个二肽,基本点是将一个氨基酸的羧基与另一个氨基酸的氨基连结成肽键,示意如下:
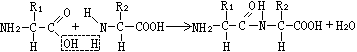 这是一个缩合反应,脱去一分子水。关键在于需要有措施使第1个氨基酸的氨基与第2个氨基酸的羧基不参与反应。方法是将这些不希望参加反应的基团用一些特殊的化学试剂与之反应,使生成的基团对缩合反应不敏感,还能在肽合成后,在不破坏肽键及氨基酸结构的条件下将它们恢复成原先的基团。这一类试剂称保护试剂,形成的基团称保护基。两个小肽缩合成较大的肽的原则也同此。因此肽的化学合成可归结为两个问题,一是不希望参与肽键缩合反应的基团的保护,二是用缩合剂引起缩合反应。
这是一个缩合反应,脱去一分子水。关键在于需要有措施使第1个氨基酸的氨基与第2个氨基酸的羧基不参与反应。方法是将这些不希望参加反应的基团用一些特殊的化学试剂与之反应,使生成的基团对缩合反应不敏感,还能在肽合成后,在不破坏肽键及氨基酸结构的条件下将它们恢复成原先的基团。这一类试剂称保护试剂,形成的基团称保护基。两个小肽缩合成较大的肽的原则也同此。因此肽的化学合成可归结为两个问题,一是不希望参与肽键缩合反应的基团的保护,二是用缩合剂引起缩合反应。在氨基保护方面,Cbz-和Boc-可以同时分别保护α-和ε-氨基是经典使用的,现更普遍采用的氨基保护基团是能轻微酸解去除的联苯异丙氧羰酰基-(BPOC-)和能被有机弱碱去除的芴甲氧羰酰基-(Fmoc-)。它们均适宜在侧链以Boc-保护的肽的合成中使用。常用的羧基保护基团有甲酯、乙酯、苄酯和叔丁酯。固相合成中固体颗粒多以苄酯与氨基酸羧基相接,此时,叔丁酯(-OtBu)则往往用于保护侧链羧基。除了侧链氨基和羧基外,组氨酸的咪唑基、精氨酸的胍基、半胱氨酸的巯基,以及酪氨酸的酚基和丝、苏氨酸的羟基等侧链基团,均应在合成肽前先行保护。
缩合反应可以以合成 Ala-Leu-Gly-Val(见氨基酸)四肽为例,示意如下(Boc、Cbz及OtBu为保护基):
Leu Gly
↓(1) ↓(2)
Cbz·Leu·OH H·Gly·OC2H5
↓(3)
Cbz·Leu·Gly·OC2H5 Val
↓(4) ↓(2)
Cbz·Leu·Gly·OH H·Val·OtBu
↓(5)
Ala Cbz·Leu·Gly·Val·OtBu
↓(1) ↓(6)
Boc·Ala·OH H·Leu·Gly·Val·OtBu
↓(7)
Boc·Ala·Leu·Gly·Val·OtBu
↓(8)
Ala·Leu·Gly·Val
整个过程可分解成下列步骤:①氨基保护;②羧基保护;③缩合;④羧基脱除保护(皂化);⑤缩合,向C端延长;⑥氨基脱除保护(催化氢解);⑦缩合,从N端延伸;⑧氨基及羧基保护同时脱除,形成无保护的四肽(轻微酸解)。
上述反应都在溶剂中进行,称为溶液方法。因所用保护基都是疏水的,而肽键是亲水的,因此合成到一、二十肽时,使保护了的肽溶解便成了困难的问题。寻找合适的溶剂是液相法的一个难题。
1963年,R.B.梅里菲尔德提出固相合成的方法,避免了溶解问题。它的原理是将第 1个氨基酸接到不溶的固体颗粒上,然后将保护的第2个氨基酸与之缩合。产物因在固体颗粒上,因此很容易将反应中剩余的试剂清洗干净。然后脱去保护基,再将第3个氨基酸与之缩合而成三肽。如此反复,只要反应产率非常高,就能合成很大的肽。最后将合成的肽从固体的支持物上切下,再经纯化,即可得相应的肽。反应可示意如下:
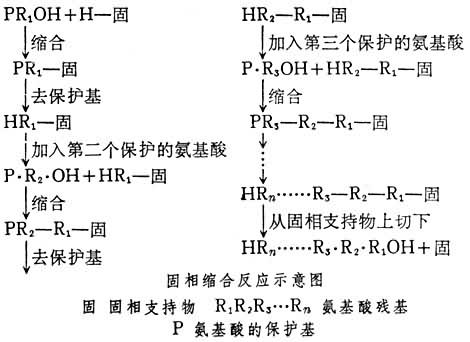
自70年代后半期始,这一方法已变成合成中等大小的肽的常用方法。
成就 50年代初期,V.迪维尼奥提纯了催产素和加压素,测定了它们的结构,并在1953年合成了第1个多肽──催产素。中国科学家在1960年成功地将天然胰岛素拆分为两条肽链,并重新组合成结晶胰岛素。同时合成了胰岛素A及B链的许多肽段,1965年合成出A链(21肽)及B链(30肽)并组合成结晶牛胰岛素。这是第1次人工合成蛋白质。R.B.梅里菲尔德在1963年提出固相合成方法,经改进,已设计出合成多肽的自动装置。他因此成就获1984年诺贝尔化学奖。