reaction mechanism
Introduction
in chemical reactions (chemical reaction), the detailed processes by which chemical substances are transformed into other substances. The reactions themselves may involve the interactions of atoms (atom), molecules (molecule), ions (ion), electrons (electron), and free radicals, and they may take place in gases (gas), liquids (liquid), or solids (solid)—or at interfaces (interface) between any of these.
The study of the detailed processes of reaction mechanisms is important for many reasons, including the help it gives in understanding and controlling chemical reactions. Many reactions of great commercial importance can proceed by more than one reaction path; knowledge of the reaction mechanisms involved may make it possible to choose reaction conditions favouring one path over another, thereby giving maximum amounts of desired products and minimum amounts of undesired products. Furthermore, on the basis of reaction mechanisms, it is sometimes possible to find correlations between systems not otherwise obviously related. The ability to draw such analogies frequently makes it possible to predict the course of untried reactions. Finally, detailed information about reaction mechanisms permits unification and understanding of large bodies of otherwise unrelated phenomena, a matter of great importance in the theory and practice of chemistry.
Generally, the chemical reactions whose mechanisms are of interest to chemists are those that occur in solution and involve the breaking and reforming of covalent bonds (covalent bond) between atoms—covalent bonds being those in which electrons are shared between atoms. Interest in these reactions is especially great because they are the reactions by which such materials as plastics (plastic), dyes (dye), synthetic fibres (fibre, man-made), and medicinal agents are prepared and because most of the biochemical reactions of living systems are of this type. In addition, reactions of this kind generally occur in timescales convenient for study, neither too fast nor too slow, and under conditions that are easily manipulated for experimental purposes. There are a number of techniques by which the mechanisms of such reactions can be investigated.
Chemical reactions involve changes in bonding patterns of molecules—that is, changes in the relative positions of atoms in and among molecules, as well as shifts in the electrons that hold the atoms together in chemical bonds. Reaction mechanisms, therefore, must include descriptions of these movements with regard to spatial change and also with regard to time. The overall route of change is called the course of the reaction, and the detailed process by which the change occurs is referred to as the reaction path or pathway.
Also important to the study of reaction mechanisms are the energy requirements of the reactions. Most reactions of mechanistic interest are activated processes—that is, processes that must have a supply of energy before they can occur. The energy is consumed in carrying the starting material of the reaction over an energy barrier. This process occurs when the starting material absorbs energy and is converted to an activated complex or transition state. The activated complex then proceeds to furnish the product of the reaction without further input of energy—often, in fact, with a release of energy. Such considerations are important to an understanding of reaction mechanisms because the actual course that any reaction follows is the one that requires the least energy of activation. This reaction course is not always the one that would seem simplest to the chemist without detailed study of the different possible mechanisms.
The study of reaction mechanisms is complicated by the reversibility of most reactions (the tendency of the reaction products to revert to the starting materials) and by the existence of competing reactions (reactions that convert the starting material to something other than the desired products). Another complicating factor is the fact that many reactions occur in stages in which intermediate products (intermediates) are formed and then converted by further reactions to the final products. In examining chemical reactions, it is useful to consider several general subjects: (1) factors that influence the course of chemical reactions, (2) energy changes involved in the course of a typical reaction, (3) factors that reveal the mechanism of a reaction, and (4) the classification of reaction mechanisms. With this information in mind, it is then possible to look briefly at some of the more important classes of reaction mechanisms. (The articles acid-base reaction (acid–base reaction), oxidation-reduction reaction (oxidation–reduction reaction), and electrochemical reaction deal with the mechanisms of reactions not described in this article.)
General considerations
Determinants of the course of reaction
The reactants
In analyzing the mechanism of a reaction, account must be taken of all the factors that influence its course. After the bulk chemical constituents have been identified by ordinary methods of structure determination and analysis, any prereaction changes involving the reactants, either individually or together, must be investigated. Thus, in the cleavage of the substance ethyl acetate by water ( hydrolysis), the actual reagent that attacks the ethyl acetate molecule may be the water molecule itself, or it may be the hydroxide ion (OH−) produced from it.
The hydrolysis of ethyl acetate can be represented by the following equation:

in which the structures of the molecules are represented schematically by their structural formulas. An arrow is used to indicate the reaction, with the formulas for the starting materials on the left and those of the products on the right. In the structural formulas, the atoms (atom) of the elements are represented by their chemical symbols (C for carbon, H for hydrogen, and O for oxygen), and the numbers of the atoms in particular groups are designated by numeral subscripts. The chemical bonds of greatest interest are represented by short lines between the symbols of the atoms connected by the bonds.
Important to this reaction is an equilibrium involving the cleavage of the water molecules into positively and negatively charged particles (ions), as follows:

In this equation the numeral in front of the symbol for the water molecule indicates the number of molecules involved in the reaction. The composite arrow indicates that the reaction can proceed in either direction, starting material being converted to products and vice versa. In practice, both reactions occur together, and a balance, or equilibrium, of starting materials and products is set up. The significance of this equilibrium for the hydrolysis of ethyl acetate is that any of the three entities (water molecules, hydronium, or hydroxide ions) may be involved in the reaction, and the mechanism is not known until it is established which of these is the actual participant. This often can be established if it is possible to determine the relative amounts of the three in the reaction medium and if it can be shown that the rate of the reaction depends upon the amount (or concentration) of one of them. Under certain conditions the hydrolysis of ethyl acetate is found to involve water molecules (as shown in the equation above); in other cases, hydroxide ion is involved.
The transition state (transition-state theory)
The transition state, or activated complex, is the fleeting molecular configuration that exists at the top of the energy barrier that the reactants must surmount to become the products. It is not strictly a component of the reaction system, and it cannot be examined directly in the way that an intermediate (however unstable) can, because it lasts no longer than the duration of a molecular collision. The transition state may have properties of its own, not reflected in those of the starting materials or of the products and of the reaction, and so it is of vital importance in determining the course of reaction. Inference concerning the nature of the transition state is the essence of mechanistic study.
The solvent
The solvent, or medium in which the reaction occurs, may perform the mechanical—but often vital—role of allowing otherwise immiscible reactants to come together rapidly. Among the important groups of solvents, each with its own special type of behaviour, are hydroxylic solvents (the molecules of which contain hydroxyl 【−OH】 groups, such as water and alcohols (alcohol)), dipolar aprotic solvents (the molecules of which show a separation of electrical charge but do not easily give up a proton, or positive hydrogen ion; e.g., acetone), and nonpolar solvents (the molecules of which do not show charge separation; e.g., hexane).
In dissolving the reactants, the solvent may interact with any or all of them, and it may be involved in the transition state for any reaction available for the system. If the solvent interacts more powerfully with the transition state than with the reactants, it facilitates the reaction. The solvent itself may of course be one of the reactants, and this circumstance introduces special problems because of the difficulty of distinguishing experimentally between its functions as a reagent and as an environment for the reaction.
Catalysts
Catalysts (catalyst) are substances that speed up a reaction by facilitating a particular mechanism—sometimes by influencing an existing prereaction and sometimes by making a new process energetically favourable. Their presence or absence frequently determines the course a reaction may take, simply because one of a number of competing reactions is, or is not, favoured. (Most catalysts are changed chemically while they speed up a reaction; sometimes—but not always—they are consumed, and sometimes they are reformed and so appear to be unchanged in concentration during a reaction.)
The products
All reactions are reversible in principle, and the nature of the products of the reaction can affect the reaction course in a number of ways. When the position of equilibrium is unfavourable, for example, the accumulation of products may cause a reversal of the reaction. In such circumstances, the physical removal of the products (through their volatility or insolubility, for example) facilitates the completion of the forward process. Sometimes too, one of the products acts as a catalyst or as an inhibitor, behaviour that strongly influences the course of the reaction.
The reaction conditions
The conditions under which some reaction occurs, including such variables as the temperature and concentrations of reactants, also are important in determining the course of the reaction. For reactions that have a high energy barrier between reactants and products, the rate is highly responsive to change in temperature, and such reactions become more likely at increased temperatures, so the minor products of a reaction often appear in larger proportions at higher temperatures.
Similarly, the concentration of reagents can be important to the course of a reaction, especially if two mechanisms are available that involve different numbers of molecules in the transition states. Higher concentrations of a particular reagent favour those mechanisms in which greater numbers of molecules are involved in the transition state. The pressure applied to the reacting system also may be significant, partly because it has an effect on concentration and partly because mechanisms involving closely associated transition states become more favourable at high pressures. The latter relationship comes about because associated transition states are those in which several molecules or ions are brought close together (and therefore take up less space), a situation that is encouraged by increased pressures.
Energy changes involved in reactions
Collisions between molecules are rapid; therefore, reactions that occur spontaneously whenever the reagents collide are fast at ordinary concentrations. However, a reaction may be restricted in rate by its dependence on the occurrence of molecular collisions, because, for example, the reagents are present in such small amounts that reactions can only occur when they happen to encounter one another. Such a reaction is said to be diffusion-controlled, because it is dependent on the process of diffusion to bring the molecules together. In such cases the viscosity of the medium is relevant; the more viscous, or “thick,” the medium, the more difficult the diffusion and the slower the reaction.
 However, most reactions involve a rate-limiting energy barrier, and it is the nature of this barrier and of the molecular configuration at its top that determines the mechanism. Diagrams of energy changes during the course of reaction often are used to illustrate the energetic aspects of the reaction. An example of a possible energy diagram for a hypothetical one-stage process—the dissociation in a solution of a covalent molecule designated E–N, into its ions, E+ and N−—is shown in the figure-->.
However, most reactions involve a rate-limiting energy barrier, and it is the nature of this barrier and of the molecular configuration at its top that determines the mechanism. Diagrams of energy changes during the course of reaction often are used to illustrate the energetic aspects of the reaction. An example of a possible energy diagram for a hypothetical one-stage process—the dissociation in a solution of a covalent molecule designated E–N, into its ions, E+ and N−—is shown in the figure-->.In this diagram the energy is plotted against a reaction coordinate—a spatial relationship that varies smoothly during the course of the reaction—in this case the distance between the portions of the molecule designated E and N. At the left-hand energy minimum of the figure, the bond between E and N is fully formed; if energy is applied to excite the system in such a way that E and N are brought even closer together (region a of the figure), the atomic nuclei (nucleus) repel, with the result that energy rises steeply. Alternatively, excitation energy, such as thermal energy from collisions with other molecules, may stretch the bond, and the energy curve then moves into region b. The E–N bond is thereby weakened steadily until the transition state is reached. This point, as can be seen in the figure, has an energy peak on the reaction coordinate. At the same time, this point represents the minimum energy required to convert the reactants into the products. The curve shown should be considered as only a planar section of a three-dimensional energy surface relating to the various possible spatial relations between the components of the reaction. The passage of the reactants from the initial state to the products then can be thought of as analogous to the climb from a valley (the initial state) through the lowest mountain pass leading to a second valley (the products). Thus, although the transition state represents a peak on the single curve depicted, it really represents a secondary minimum (or pass) in the energy surface. From the top of this pass (the transition state), the molecule can only descend, losing energy by collision. In doing so it may revert to the starting materials, or it may dissociate to give the products (region c in the figure). The products in the case of the reaction chosen are the ions E+ and N−, which are held together by electrostatic attraction as an ion pair at the right-hand minimum of the graph; beyond this point, further separation of the ions involves the consumption of energy (region d). In principle, the products may lie at higher or (as shown) lower energy than that of the initial state. The mechanism of a reaction such as this may be considered to be completely defined when the structures and energy properties of the starting materials, the products, and the transition state are known.
Ultimately, it should become possible to compute the properties of the molecules solely from the properties of their constituent atoms and also to deduce the transition states for any of the reactions these molecules may undergo. For a few simple situations, approaches already have been made to definitions of mechanism in this degree of detail. Systems involving several atoms, however, require a many-dimensional representation of the reaction course instead of the two-dimensional description shown in the figure. The problems of computation, and of testing theory against experiment, then become enormous. Nonetheless, attempts have been made to deal with some simple reactions of systems involving up to about five atoms.
 For a reaction involving several distinct stages, a more complicated description of the reaction course also is necessary. The figure--> gives an example of such a situation. This hypothetical reaction is reversible, with three successive intermediate complexes formed between the reactants. Unlike the case of the simpler situation above, the physical process that best approximates what is happening along the reaction coordinate changes from stage to stage across the diagram.
For a reaction involving several distinct stages, a more complicated description of the reaction course also is necessary. The figure--> gives an example of such a situation. This hypothetical reaction is reversible, with three successive intermediate complexes formed between the reactants. Unlike the case of the simpler situation above, the physical process that best approximates what is happening along the reaction coordinate changes from stage to stage across the diagram.The highest point on the energy diagram corresponds to the energy of the transition state of the rate-limiting step in the reaction—that is, the slowest step in the reaction, the one that governs, or limits, the overall rate. The rate of reaction is independent of the nature and number of the intermediates that lie before this transition state on the reaction coordinate. Progress along the reaction coordinate cannot be identified with the time course of the reaction because any individual pair of reactants may reside for an appreciable time in the partially activated state represented by one of the intermediate complexes before final reaction is achieved. Once the reactants have passed the rate-limiting transition state, they must lose energy (usually by collision with other molecules) to reach the final state. In a sense, the reaction coordinate in such a reaction may be thought of as representing the chemical course of the reaction (rather than a spatial or a time course). If the diagram represents the only reaction of the system, then it is possible to apply the principle of microscopic reversibility, which states that the course taken by reverse action will be statistically identical with that taken by the forward reaction. This principle is not applicable to a reaction giving several different products not at equilibrium with one another, however.
Reaction mechanisms: nature of reactants, intermediates, and products
The stoichiometry of a reaction consists of the chemical formulas and relative molecular proportions of starting materials and products. Obviously these have a bearing on the mechanism of the reaction, for the overall reaction course must proceed from starting materials to the products. The stoichiometry of the reaction may be misleading, however, because the participants in the overall reaction may not be involved directly in the rate-limiting step.
The discovery of intermediates in the course of a reaction is important because these point to the existence of distinct stages, the mechanism of each of which must then be determined. The identification of intermediates that persist only briefly or that are present in only small amounts depends on the availability of powerful, sensitive, and rapid experimental techniques. For this purpose, a number of specialized instrumental procedures (including ultraviolet, infrared, magnetic resonance, and mass spectrometry) are widely used to supplement the more usual chemical and physical methods.
The identification of a new chemical substance formed transiently in a reaction mixture, however, does not unambiguously imply that that substance is an intermediate in the reaction. In many cases a newly found material is only a temporary repository of the proportion of the reactants and ultimately produces the products by first reverting to the starting material.
The identification of the products of a reaction also helps to define the reaction course, because the mechanism in question clearly must account for their formation. Mechanistic theory has been greatly facilitated by the development of powerful methods of separation and purification based on chromatography (separation of compounds on the basis of their relative degrees of adsorption to certain solid substances, such as starch or silica) and also by modern methods available for the analysis of small quantities of materials. These spectroscopic procedures are often used, as is another instrumental method called polarimetry.
An important consideration with regard to the products of the reaction is whether the reaction is under kinetic or thermodynamic control. A reaction is said to be kinetically, rather than thermodynamically, controlled when the products are formed in proportions different from those that would prevail at equilibrium between the same products under the same conditions. Thermodynamic control leads to the equilibrium ratio of the products. Often, though not invariably, reactions under kinetic control give a greater amount of the thermodynamically less stable of two possible products; if thermodynamic control is then established, the products shift to their equilibrium proportions, which might give a misleading picture of the reaction course. Hence, inferences concerning the nature of the transition state can be drawn from the nature of the products only with a good deal of circumspection.
In determining the mechanism of a reaction, one of the major problems is to deduce the spatial or three-dimensional changes that occur to the molecules involved as they proceed from their initial state through the intermediate stages and transition states to the final products. Knowledge about such changes can generally be deduced from knowledge of the stereochemistry (three-dimensional structures) of the starting materials, intermediates, and final products (provided these are obtained under kinetic control). Information of this kind is obtained by determinations of optical activity and analysis of the structures of the compounds by standard means.
In certain instances, information about the movement of atoms between molecules during the course of a reaction can be gained by using compounds containing isotopes of certain of the atoms. These isotopes behave much like the ordinary atoms they replace, but they can be identified by their behaviour. For example, in the hydrolysis of ethyl acetate (see above The reactants (reaction mechanism)), it is crucial to a determination of the mechanism to be able to establish which of the two reactants (ethyl acetate or water) provides the oxygen atom that ends up in the product ethyl alcohol. In this case, the use of water labelled with oxygen-18 reveals that the oxygen atom in the alcohol comes from the ethyl acetate molecule.
Effects of reaction conditions and environment
Kinetic order
Because the possibilities that need to be considered for the transition state have been limited by determination of the chemical structures of the participants, the most powerful method of obtaining further information is the use of the kinetic method—i.e., the study of the effect of reaction conditions on the rate of reaction. Experimental methods that have been used in kinetic studies include most of the known methods of chemical separation and analysis. Techniques that involve removing samples from the reaction mixture at intervals or stopping the reaction and analyzing for starting material or product are common for reactions with half-lives down to about a minute. For faster reactions, methods involving rapid scanning and automatic recording of some characteristic property of the reacting mixture, such as absorption of light at a particular wavelength, recently have become important. Other procedures for following exceptionally fast reactions include the controlled supply of a reagent within extremely small concentration limits, sometimes by electrolytic procedures (the use of an electric current to produce precise amounts of the substance) and sometimes by carrying out the reaction under conditions in which separate flowing streams of the reactants come together to ensure rapid mixing. So-called relaxation procedures—in which a system in equilibrium is very rapidly perturbed and its rate of relaxation to the original or to some new equilibrium state is observed—also have been applied to the study of reactions of extremely small half-lives.
The mechanistic information to be obtained from the observed kinetic behaviour of a system derives from the fact that, for an activated process, the transition state can be considered to be in thermodynamic equilibrium with the starting materials except with respect to its motion along the reaction coordinate. It follows that the rate of reaction is approximately proportional to the product of the concentrations of those substances that comprise the transition state. If the concentrations of all but one reactant are held constant while the concentrations of that reactant are changed, then the variation in rate with the concentration changes will establish how many molecules of that particular reactant are involved in the transition state. This figure is called the order of reaction with respect to the reactant in question. A full description of the composition of the transition state then requires identification of the order of reaction with respect to each of the reactants.
Although straightforward in principle, the application of this method involves some difficulties. Sometimes (for example, with ionic solutes in solvents of low polarity) the concentrations of the reagents are not truly representative of their influence on the reaction rate. The kinetic order then does not properly represent the composition of the transition state. The power of the kinetic method is often greatly increased when the rate of reaction can be followed by more than one method. In such instances, it frequently has been found that unexpected differences reveal the intervention of previously unsuspected intermediates.
Environmental effects
Changes in the environment (such as the composition of the solvent) frequently influence the course of the reaction by affecting the relative stabilization of the initial state and the transition state. Large changes in the polar character (charge distribution) of the solvent, for example, may have an effect on the course of the reaction if there is a substantial change in polarity between the reactants and the transition state.
Electronic and isotopic effects
Structure-reactivity relations
An observed correlation of changes in a reaction rate with systematic changes in the structure of one of the reactants often reveals the movements of electrons between atoms as the reactants shift toward the transition state. Systematic changes in structure usually are brought about by selecting a particular molecular system and varying a portion of it (such as, for example, the substituents on a benzene ring). The effects of each variant on the rates of several different reactions are determined experimentally, and the results are plotted on graphs; the values resulting from a particular molecular variation in one reaction are measured along one axis and those of the same variation in the other reaction along the other axis. A straight-line relationship indicates that the molecular changes are affecting the rates of the two reactions in related ways. The slope of the line gives a comparison of the relative response of the two systems to the given change in structure; the sign of the slope tells whether a particular structural change favours both reactions or favours one while disfavouring the other. The observed effects generally can be correlated with the electronic nature of the molecular variants introduced. For example, if a substituent in the molecule tends to donate electrons toward the reactive centre in the molecule and this change favours the reaction, it can be concluded that an electron-rich centre is involved in the transition state.
Electronic effects of the above kinds can be complicated by spatial, or steric, factors. (A reaction is said to be sterically hindered when the transition state is more congested than the initial state and to be sterically accelerated when the reverse is true.) When suitable allowance is made for the above electronic influences, structural changes can be used to help define the detailed geometry of the transition state. Thus, if large or bulky substituents have an inhibiting effect on the course of the reaction, it can be concluded that the transition state differs from the starting material in such a way that the effect of the bulky group is accentuated; in the alternative situation, in which a bulky substituent accelerates the reaction, it may be concluded that the formation of the transition state relieves crowding found in the starting material. Although absolute calculations of reactivity—that is, calculations based on molecular structure alone—have made little progress in the case of polyatomic systems, significant calculations of reactivity differences have been made in favourable instances in which steric and electronic effects can be disentangled.
Kinetic isotope effects
Isotopes (isotope) are atoms that have the same atomic number (and, hence, generally the same chemistry) but different mass. The difference in mass becomes chemically important in certain instances. For example, when a carbon-hydrogen bond is replaced by a carbon-deuterium bond (deuterium being an isotope of hydrogen with about twice the mass), the vibrational frequencies of that bond are changed. The vibrational stretching frequency of a bond between two atoms, for example, gives an approximate measure of the bonding forces holding those two atoms together, the effective masses of the two atoms being allowed for. If the character of the carbon-hydrogen bond is altered between the normal state and the transition state, the change from hydrogen to deuterium may have an effect on the relative stabilities of the normal and transition states and also, therefore, on the rate of reaction. These effects of isotopic substitution are called kinetic isotope effects. In cases in which the atom that is substituted is linked to the rest of the molecule by only one bond, the bond involving the heavier isotope is usually more difficult to break than the one involving the lighter isotope. Isotope effects are large only for the isotopes of hydrogen, but, with heavier elements, even small differences can give important information about the mechanism, provided that sufficiently precise methods are available for their measurement.
Classification of reaction mechanisms
There is no one generally agreed-upon and completely satisfactory method of classifying mechanisms; individual authors have often adopted their own nomenclature and symbolism. There are, however, a number of useful classification principles that should be noted.
Homolysis and heterolysis
When a covalent bond (a nonionic chemical bond formed by shared electrons) is made up of two electrons, each of which is supplied by a different atom, the process is called colligation; the reverse process, in which the electrons of a covalent bond are split between two atoms, is known as homolysis. These reactions are shown schematically by the equation

in which A and B represent the separate atoms (or groups), the single dots represent electrons, and the double dots represent the electron pair that makes up the bond. The products of a homolysis reaction are called free radicals, and all such processes are said to have homolytic or free-radical mechanisms.
If, on the other hand, a covalent bond is formed by a pair of electrons both of which come from one of the two reagents, the process may be described as coordination and its reverse as heterolysis. Coordination and heterolysis are shown schematically by the equation

in which the dots indicate the electron pair and the letters N and E represent the atoms (or groups) that, respectively, donate and accept the electrons (see below for special significance of the letters N and E). Reactions of this kind are said to have heterolytic mechanisms.
Nucleophilicity (nucleophile) and electrophilicity (electrophile)
In a heterolytic reaction, the unit that carries the electron pair (designated N) is nucleophilic; i.e., it seeks an atomic nucleus to combine with. Conversely, the other unit in the reaction (designated E) is electrophilic; it seeks to combine with a pair of electrons. An electrophilic reaction mechanism is one that involves an electrophilic reagent attacking a nucleophilic substrate. Because every such reaction involves both an electrophilic substance and a nucleophilic substance, it must be agreed arbitrarily which unit is the reagent and which the substrate. Often this agreement is made on the basis of molecular size, the larger-sized material being classed as the substrate.
In certain reactions in which movements of electrons are concerted and cyclic, it is not possible to identify any one reagent—or even any one particular atom or set of atoms in the molecule—as electrophilic or nucleophilic. Such processes are classified as electrocyclic.
Molecularity
The mechanism of an individual stage of a reaction can be described as unimolecular, bimolecular, and so on, according to the number of molecules necessarily concerned in covalency change in the transition state. As an extension of this classification, the number of molecules involved in the rate-limiting step of a several-stage reaction also can be used for classification of the overall reaction. Ambiguities and blurred distinctions arise when there are strong interactions between the solvent and the initial or transition state or when two stages of a reaction are so near in rate that they become jointly rate-determining. Nevertheless, classifications based on molecularity are widely used.
Intermolecularity and intramolecularity
The distinction between intermolecular and intramolecular processes is often useful. In intermolecular reactions, covalency changes take place in two separate molecules; in intramolecular reactions, two or more reaction sites within the same molecule are involved.
Nature of catalysis
Classification also can be made on the basis of the mode of catalytic action. In ester hydrolysis, as in the hydrolysis of ethyl acetate (see above The reactants (reaction mechanism)), for example, distinction can be made between mechanisms in which catalysis is brought about by protons (proton) (hydrogen ions) or by acids (acid) in general, by hydroxide ion or by bases (base) in general, or by enzymes.
Kinetic order
The kinetic order of a reaction is best considered as an experimental quantity related to (but not identical with) the number of molecules of any reactant involved in the transition state. It may for various reasons reveal only a part of what is happening in the rate-limiting transition state, one reason being that the concentrations of the components of the transition state may not all change with progress of the reaction. Establishing that a reaction is of the first order kinetically (that is, behaves as though only one molecule of the reactant is involved in the transition state) with respect to one of the components, for example, seldom reveals whether or not one or more molecules of solvent also is involved in the transition state. This is so because the solvent is present in such large amounts that its concentration does not change effectively with the course of the reaction. For this reason, the order with respect to an individual component may sometimes be useful for classification, but, for the overall reaction, molecularity is the more fundamental quantity.
Time sequence of events
The time sequence of events in a chemical reaction also provides a means of classification. In some mechanisms the bond-making and bond-breaking processes occur together and are said to be concerted; in others the individual stages are discrete, with recognizable intermediates occurring between them, and it may be necessary to specify not only that the mechanism is stepwise but also the order in which the steps occur.
Comparison of selected reaction mechanisms
For the following incomplete and abbreviated survey of reaction mechanisms, several mechanisms important in the development of mechanistic study have been chosen.
Nucleophilic substitutions at saturated carbon centres
The term substitution refers in general to the replacement of any group in a molecule by any other group. Saturated carbon centres are carbon atoms at which no multiple bonds occur, and nucleophilic substitutions—those brought about by nucleus-seeking reagents—can occur at such carbon atoms (atom) by either of two main mechanisms: bimolecular and unimolecular.
Bimolecular
In bimolecular nucleophilic substitution reactions in which the substrate is attacked at a saturated carbon atom, the starting material has a tetrahedral structure, and the transition state has a trigonal bipyramidal structure (both of which are shown below). Each individual act of substitution produces a product of inverted (i.e., mirror-image) stereochemical configuration.
A typical bimolecular substitution reaction is shown by the equation

in which the chemical symbols represent atoms of the elements as above (with Br the symbol for an atom of bromine and N the symbol for any nucleophilic agent). This equation differs from the earlier ones in that a three-dimensional representation of the structures is intended. The three-dimensional effect is achieved by considering that the bonds represented by ordinary solid lines lie in the plane of the paper, bonds represented by dashed lines project to the rear, and bonds represented by dark triangles project to the front. A further unique feature of this equation is that the representation of the transition state shows half bonds (bonds in the process of being formed or broken), which are indicated by dotted lines. In addition, in the transition state, half negative charges are indicated by the symbols “1/2−.” The mechanism of this reaction is characterized by entry of the nucleophilic reagent from one side of the substrate molecule and departure of the bromide ion from the other side. The resulting change in configuration of the substrate has been likened to the turning inside out of an umbrella, with the transition state representing that precise moment when the ribs are essentially vertical in the course of their passage from one side of the structure to the other. The reaction is synchronized, or synchronous, in that entry of the nucleophile and departure of the leaving group occur simultaneously. It is bimolecular in that one molecule each of substrate and nucleophile are involved in the transition state, and it is stereospecific in that the stereochemical outcome of the reaction is invariably the same.
This bimolecular mechanism occurs with a wide range of structures. It often can be characterized by second-order kinetics—i.e., by reaction rates that are dependent on the concentrations of both the substrate and the nucleophilic reagent. The transition state is highly congested, so that effects of steric hindrance are large. Otherwise, however, structural changes produce a variable response because of the conflicting electronic requirements of the bond-forming and bond-breaking processes. Bimolecular nucleophilic substitutions with rearrangement of the bonding skeleton also are known.
Unimolecular
Unimolecular nucleophilic substitution reactions proceed by a two-stage mechanism in which heterolysis precedes reaction with the nucleophile. The following equation is a typical example:

in which the symbols are the same as in earlier equations, with the addition of delta plus (δ+) and delta minus (δ−), which indicate partial positive and negative charges, respectively. The significant consideration in this reaction mechanism is the initial separation of the bromide ion (by way of a transition state showing partial separation of the ion) to give a free positively charged organic ion ( carbonium ion). This step is the rate-determining step of the reaction, and, because it involves only a molecule of the substrate, the reaction is unimolecular. The second stage of the reaction is the interaction of the intermediate carbonium ion with the nucleophile to give the products of the reaction.
The unimolecular reaction is characterized experimentally by first-order kinetics—i.e., by a rate that depends only on concentration of the substrate (and not the nucleophile), by the absence of effects of steric hindrance, by powerful facilitation of the reaction by the presence of electron-releasing groups attached to the reaction centre, and by variable, and often diagnostic, stereochemistry. Inversion of stereochemical configuration (change from one configuration to the mirror-image configuration) is frequently encountered, accompanied by racemization (production of both mirror images). The extent of racemization depends upon the life of the intermediate carbonium ion, with longer-lived ions leading to more extensive racemization (due to the fact that the symmetrical ion is exposed to attack from either side).
In an important group of structures, a group not formally involved in the overall reaction interacts with a carbonium ion centre to form an intermediate, which then reacts with the nucleophile to give a product of the same stereochemical configuration as the starting material. This behaviour can be represented by the equation

In the first demonstrations of this behaviour, the participating group (G) was a carboxylate anion group, which can be represented in chemical symbols as

Subsequent investigations revealed numerous examples involving other substituents, and the phenomenon is now commonly described as neighbouring-group participation.
A frequent consequence of reaction through intermediates having carbonium ionic character is that some of the products have rearranged skeletal structures. In this equation the symbol Cl represents a chlorine atom.
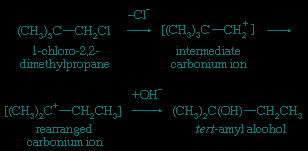
The fundamental difference between the transition states in the bimolecular and unimolecular mechanisms is the degree of covalent bonding between the nucleophile and the substrate in the transition state. In the unimolecular mechanism such bonding is negligible; in the bimolecular case it has essentially reached the half-bond status. In borderline situations the matter is difficult to resolve, a number of intermediate cases being known, and there has been much controversy as to the validity of the distinction between the bimolecular and the unimolecular mechanisms. Experimentally, however, clear examples of each class have been established.
Nucleophilic substitution at unsaturated carbon centres
Unsaturated carbon centres—including those involving ordinary carbon-carbon double bonds and those involving the extended cyclic systems of alternate single and double bonds known as aromatic rings—are not easily attacked by nucleophilic reagents unless they have been denuded of electrons by electron-attracting substituents. A two-stage process that includes addition of the nucleophile followed by expulsion of a negatively charged (anionic) group is the course normally taken for substitutions at aromatic centres. The presence of the aromatic ring enforces the geometry of the product, and the reaction is favoured by electron-withdrawing groups, such as the nitro (−NO2) group, which help to accommodate the negative charge on the intermediate. An example of this type of reaction is the displacement of fluoride ion from 2,4-dinitrofluorobenzene by nucleophiles such as ethoxide ion.
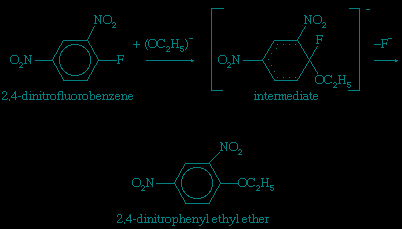
In this equation fluorine atoms are indicated by the chemical symbol F; nitro groups (consisting of one nitrogen and two oxygen atoms) are indicated by the symbols −NO2; normal benzene rings (of six carbon atoms, each of which carries a single hydrogen atom) are indicated by regular hexagons with circles in them; and benzene rings containing disrupted electronic structures are indicated by hexagons with partial dotted circles.
Substitution reactions at ordinary double bonds (olefinic bonds) also take place by a two-stage process. When the two stages in the reaction occur synchronously or in very quick succession, the product has the same geometrical relationship that existed in the starting material. If, however, the anionic intermediate has sufficient lifetime, rotation about the new carbon-carbon single bond can precede loss of the negatively charged group, resulting in production of two products of differing molecular geometry—that is, products in which the substituents are differently situated with respect to the double bond.

If the intermediate anion takes up a hydrogen ion (proton) and then loses hydrogen and halogen simultaneously (concerted elimination), the reaction is then said to be following an addition-elimination sequence. Examples of such reactions are known, particularly in situations in which the double bond includes an atom other than carbon. In aromatic systems the reverse situation, in which elimination occurs, followed by addition, also is found. Finally, unimolecular mechanisms of substitution also are known to take place at particularly activated unsaturated centres. For example:

in which the symbol Ar represents a benzene ring or other aromatic system.
Electrophilic substitution at unsaturated carbon centres
Because of its wide applicability, particularly to aromatic systems, electrophilic substitution is an important reaction. Reaction by any one of several mechanisms is possible. One of the more common is shown here; reactions in this category consist of replacement of a group designated Y (often a hydrogen atom) in an aromatic molecule by an electrophilic agent designated E. Both substituents can be any one of various groups (e.g., hydrogen atoms or nitro, bromo, or tert-alkyl groups).
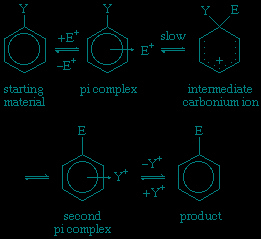
Here, Y represents a substituent on the ring; the arrow from the ring centre indicates coordination.
As shown, the reaction begins with formation of a pi complex, in which the electrons associated with the aromatic ring, or other unsaturated centres (pi electrons), coordinate weakly with the electrophile. This complex forms rapidly in an equilibrium preceding the rate-determining step, which itself leads to a carbonium ion intermediate and then by way of a second pi complex to the product. Examples are known in which the removal of the proton from the carbonium ion intermediate (to form the second pi complex) becomes rate-determining.
Reactivity by this mechanism is dominated by the electrophilic character of the reagent (E); however, it also responds powerfully to changes in structure of the organic substrate. As would be expected, substituents that release electrons toward the reaction site facilitate the reaction, and those that withdraw electrons retard reaction. These effects are very specific with regard to the position at which the modifying group is introduced.
Steric (spatial) effects generally are smaller than electronic effects in determining the characteristics of reaction by this mechanism, but they are not negligible. Direct steric hindrance and steric acceleration both have been found with suitably placed large substituents and reagents, and indirect effects arising because one group interferes with the orienting power of another also are known.
Substitution with accompanying rearrangement of the double-bond system is another established reaction path. An example is shown below in which the positions of chlorine attachment and proton loss were established by isotopic labeling.

Addition-elimination and indirect substitution reactions also can occur and are responsible for a number of unusual products formed in aromatic substitution reactions. Examples of these reaction sequences are shown below:

Addition reactions (addition reaction)
Reactions in which a multiple bond between two atoms becomes partly or fully saturated by covalent attachments at both centres are called addition reactions (addition reaction). Many mechanisms are known for such reactions; most of them are variants of four basic mechanisms, which differ chiefly in the sequence of events that occur.
With initial electrophilic attack
Addition reactions beginning with electrophilic attack include many additions to olefins (olefin) (compounds with double bonds), some additions to acetylenes (acetylene) (compounds with triple bonds), and some additions to compounds with other multiple bonds. There is a close relationship between this mode of addition and the electrophilic substitutions discussed in the preceding section, as shown by this general representation of the reaction:

in which the arrows on the olefin structure indicate the flow of electrons toward the terminal carbon, which attracts the electrophilic proton because it becomes an electron-rich centre. Electrophiles, which can be effective either as positive ions (E+) or in combination with a nucleophile (E–N), include protons (H+), carbonium ions (R3C+), positively charged halogen ions (Cl+, Br+, I+), nitronium ions (NO2+), nitrosonium ions (NO+), and many others. In general, any nucleophile can complete the reaction. When the first stage of the reaction (addition of the electrophile) is rate-determining, the rate responds powerfully to electron release to the reaction centre, and this factor determines selectively the orientation of initial attack with respect to the double bond. Thus, propylene reacts with hydrogen chloride many times faster than ethylene does, and the product is exclusively 2-chloropropane, rather than 1-chloropropane, because the concentration of electrons on the terminal carbon determines that the electrophilic proton finds it easier to attack that carbon rather than the central carbon atom.

Addition by this mechanism can be accompanied by substitution and by rearrangement as alternative reactions of the carbonium ionic intermediate. Characteristically, the ratios of product are kinetically controlled (see above Reaction mechanisms: nature of reactants, intermediates, and products (reaction mechanism)). Reactions by this mechanism can be complicated by the intervention of intermediates that are more complicated structurally. Neighbouring-group interaction can modify the structure of the intermediate toward a bridged structure and thus determine the stereochemistry of addition.
Although it is common to find that the first stage of this sequence is rate-determining, in some cases the rate-limiting transition state lies later along the reaction path. It also is possible for the two stages to be concerted, with the electrophilic and nucleophilic fragments (E and N) of the reagent E–N acting either as still covalently bound or as separate kinetic entities (E+ and N−). Especially in acid-catalyzed additions to carbon-oxygen and carbon-nitrogen double bonds, the first stage of the reaction can become rapidly reversible, and the mechanistic characteristics of the reaction are then appropriately modified.
With initial nucleophilic attack
The reverse mode of addition, in which a nucleophile initiates attack on the multiply bonded carbon atom, is less easily realized in simple systems; it does occur with acetylenes, and it also is the basis of reactions that occur when the centre of attack is denuded of electrons. For example, the formation of substances called cyanohydrins from carbonyl compounds (materials with carbon-oxygen double bonds) occurs as follows:

in which the curved arrow indicates the movement of electrons in the carbonyl group. Initial attack on carbon by the nucleophilic cyanide ion in this case is facilitated by the electron withdrawal by the oxygen atom (shown by the curved arrow in the formula). Such electron withdrawal also can be transmitted along a series of alternate double and single bonds (a conjugated system), with resultant addition to the ends of the system.
Electrocyclic
In a third class of additions, both portions of the attacking reagent combine simultaneously with the substrate. Reactions of this kind sometimes retain predominantly electrophilic or predominantly nucleophilic character, as can be shown by structural and environmental effects. In a number of important cases, however, quite different behaviour is observed. For example, the addition of cyclopentadiene to 1,4-benzoquinone follows second-order kinetics and proceeds at nearly the same rate in the gas phase and in solvents of widely differing polar character.

In this equation the polygons represent rings of carbon atoms (one at each corner), with double bonds between certain atoms as shown. Therefore, there must be little development of charge in the transition state, and the formation of the two new single bonds and the accompanying electronic movements must be well synchronized. A large number of such reactions are known; they are characterized by a remarkable stereospecificity (stereochemical specificity), controlled in part by steric effects and in part by the stereo-electronic characteristics of the combining double-bond systems.
Homolytic
Additions by free-radical mechanisms also are well known. They replace the concomitant polar additions most easily when homolytic (decomposition of a compound into two neutral atoms or radicals) fission of the reagent can be readily catalyzed and when the radicals produced as intermediates sustain chain processes. Addition of hydrogen bromide to olefins falls into this class. Equations 1–4 describe the main part of the sequence; reactions 2 and 3 are repeated many times before reaction 4 or some other reaction intervenes to break the chain. As a result, one act of initiation results in many molecules of product.

The reaction can give an orientation of substituents opposite to that found in electrophilic addition, which in the above example would produce CH3CH(Br)CH3, and in suitable cases it can be just as stereospecific.
Elimination reactions (elimination reaction)
Elimination reactions can be treated formally as the reverse of additions. The simplest examples of this class of reactions are the olefin-forming 1,2-eliminations—that is, eliminations of substituents from adjacent carbon atoms—but eliminations to give other types of double bonds are equally well known. Again, 1,3-eliminations—eliminations of substituents from carbon atoms separated by a third carbon—give compounds with three-membered rings of carbon atoms (cyclopropanes (cyclopropane)). Furthermore, the so-called conjugate eliminations occur when one or more double bonds are inserted between carbon atoms bearing the substituents that are eliminated; the result of such eliminations is a system of alternating double and single bonds (a conjugated system). Finally, there also are fragmentation reactions, in which two small fragments are lost from the organic molecule. Of these reaction types, only the 1,2-eliminations will be discussed here, it being understood that examples of the mechanisms may be found, as appropriate, in other types of elimination reactions.
Concerted, bimolecular
Concerted bimolecular eliminations are characterized by second-order kinetics; they occur readily with powerful nucleophiles. A favoured stereochemical course (trans-elimination) involves a particular geometry, as shown, which requires that in the starting material the eliminated units be situated on opposite sides of the molecule.

The olefinic product then must have the particular structure shown, rather than that of its geometric isomer. The relative extent to which the various bonds are formed and broken in the transition state varies considerably with the substrate.
Stepwise, bimolecular
If removal of the electrophilic fragment precedes the loss of the nucleophile, the reaction becomes stepwise and involves a carbanionic intermediate.

Reaction by this path, which sometimes can be characterized by exchange of protons between the solvent and the starting material, is less stereospecific than the reaction by the concerted mechanism. This lessened stereospecificity is caused by the carbanion intermediate's not maintaining the rigid geometry characteristic of the concerted mechanism.
Stepwise, unimolecular
A carbonium ion produced by heterolysis (decomposition of a compound into oppositely charged particles or ions) may lose a proton, thereby effecting a 1,2-elimination reaction:

Such eliminations, which generally accompany nucleophilic substitutions, are promoted by electron release to the carbonium ion centre. The loss of the proton usually occurs in such a way as to predominantly give the thermodynamically more stable of the alternative products.
Cyclic
Some cyclic eliminations are fully concerted, but in others the loss of a nucleophilic or of an electrophilic component can be dominant. For example, the gas-phase pyrolysis (destructive heating) of alkyl halides shows the orientation and structure effects characteristic of unimolecular stepwise elimination reactions in solution. In such cases, the transition state (shown below), though still cyclic and preserving the stereochemistry, must involve greater stretching of the carbon-chlorine than of the carbon-hydrogen bond.

Nucleophilic replacements in complexes of metals
Stable compounds with more than four groups bonded to a central atom (the situation commonly encountered in compounds of carbon) are formed by elements in the second and higher rows of the periodic table of the elements. Mechanisms of reactions of these compounds therefore become more complex on stereochemical grounds alone. Furthermore, the energy levels of electron paths (orbitals), which can accommodate the bonding electrons of the reacting atom, have become closer in these compounds, and reactions involving the formation of new bonds by expansion of the valency shell of this atom often become more readily accessible. For example, nucleophilic attack on carbon tetrachloride is slow, whereas that on silicon tetrachloride is fast, because in the former compound the attacked atom (carbon) has reached its maximum stable coordination number (indicative of the size of the valence shell) and in the latter the central atom (silicon) has not, and its valency shell can be expanded simply by the attachment of a nucleophile. A similar difference in the mechanisms of reactions of metal complexes is found, depending on whether or not the metal atoms are free to engage in valence shell expansion.
Unimolecular, in octahedral complexes
Octahedral complexes of metals of the first transition series (elements from scandium to copper) have reached their maximum stable coordination number, six. Accordingly, many of their replacement reactions are believed to occur by dissociation to give an intermediate having only five groups bonded to the reaction centre. Several different types of kinetic behaviour have been recognized. The initial stage may be a rate-determining dissociation of the cobalt complex shown below, in which methanol is the solvent, “en” is ethylene diamine (H2NCH2CH2NH2), and N− can be any of a variety of nucleophiles, including bromide, thiocyanate, and nitrate ion.

Alternatively, the dissociative stage can be a pre-equilibrium, as in many replacements of water, as shown in the reaction below.

The stereochemistry of these reaction paths is of great mechanistic significance, and it varies both with the nature of the central metal atom and the nature of the attached groups (ligands (ligand)).
Bimolecular, in square planar complexes
Square planar four-coordinated complexes differ from their octahedral six-coordinated analogues in that they generally undergo bimolecular associative, rather than dissociative, nucleophilic displacements. Thus, for many reactions involving replacement of a ligand by a nucleophile in complexes of platinum(II) ions, a kinetic effect proportional to the concentration of the nucleophile can be identified, showing that the nucleophile is involved in the transition state. Furthermore, the stereochemical specificity of such reactions as shown below can be accommodated readily in terms of the five-coordinated associated intermediate, whereas a dissociative mechanism would be expected to result in the formation of a mixture of the two geometric isomers.

A ligand across from, or trans to, a replaceable group has a much greater influence on the rate of substitution than does the same substituent next to, or cis to, the replaceable group, and this trans effect helps to define the nature of the bonding in the transition state, because it suggests that only the trans substituent is in the same plane as the associated and departing group in the intermediate.
Additional Reading
Books of general interest include Calvin D. Ritchie, Physical Organic Chemistry, 2nd ed., rev. and expanded (1990); Robert B. Jordan, Reaction Mechanisms of Inorganic and Organometallic Systems (1991); Dimitris Katakis and Gilbert Gordon, Mechanisms of Inorganic Reactions (1987); P.J. Robinson and K.A. Holbrook, Unimolecular Reactions (1972), a thorough discussion of how energy gets shuffled within a molecule to lead to reaction; Zdeněk Slanina, Contemporary Theory of Chemical Isomerism (1986), a broad survey of rearrangement mechanisms; T.L. Gilchrist and R.C. Storr, Organic Reactions and Orbital Symmetry, 2nd ed. (1979); Ralph G. Pearson, Symmetry Rules for Chemical Reactions (1976); J.N. Murrell et al., Molecular Potential Energy Functions (1984), a survey of methods and results; John W. Moore and Ralph G. Pearson, Kinetics and Mechanism, 3rd ed. (1981); James H. Espenson, Chemical Kinetics and Reactions Mechanisms (1981); Frank Wilkinson, Chemical Kinetics and Reaction Mechanisms (1980); R.P. Bell, The Proton in Chemistry, 2nd ed. (1973); E. Buncel and C.C. Lee (eds.), Isotopic Effects: Recent Developments in Theory and Experiment (1984); and Barry K. Carpenter, Determination of Organic Reaction Mechanisms (1984).Books of specialized interest include S.R. Hartshorn, Aliphatic Nucleophilic Substitution (1973); J. Milton Harris and Samuel P. McManus (eds.), Nucleophilicity (1987); Pierre Vogel, Carbocation Chemistry (1985); Herbert C. Brown and Paul von R. Schleyer, The Nonclassical Ion Problem (1977); Robert B. Bates and Craig A. Ogle, Carbanion Chemistry (1983); E. Buncel and T. Durst (eds.), Comprehensive Carbanion Chemistry (1980– ); George A. Olah, Ripudaman Malhotra, and Subhash C. Narang, Nitration: Methods and Mechanisms (1989); Kenneth Schofield, Aromatic Nitration (1980); R. Taylor, Electrophilic Aromatic Substitution (1990); and Peter B.D. De La Mare, Electrophilic Halogenation (1976).Current information may be found in the following series: Advances in Detailed Reaction Mechanisms (annual).
- Emsian Stage
- Emsland
- Ems River
- Ems telegram
- emu
- emulsifier
- emulsion
- emu-wren
- Emydidae
- enamel
- enamel miniature
- enamelwork
- enantiomorph
- enargite
- Encamp
- Encarnación
- encaustic painting
- Enceladus
- Encephalartos
- encephalitis
- enchanter's nightshade
- Enchi Fumiko
- enchondroma
- Enciclopedia italiana di scienze, lettere ed arti
- Enciclopedia universal ilustrada europeoamericana