organometallic compound
chemical compound
Introduction
 any member of a class of substances containing at least one metal-to- carbon bond in which the carbon is part of an organic group. Organometallic compounds constitute a very large group of substances that have played a major role in the development of the science of chemistry. They are used to a large extent as catalysts (catalyst) (substances that increase the rate of reactions without themselves being consumed) and as intermediates in the laboratory and in industry. The class includes such compounds as ferrocene, a remarkably stable compound in which an iron atom is sandwiched between two hydrocarbon rings.
any member of a class of substances containing at least one metal-to- carbon bond in which the carbon is part of an organic group. Organometallic compounds constitute a very large group of substances that have played a major role in the development of the science of chemistry. They are used to a large extent as catalysts (catalyst) (substances that increase the rate of reactions without themselves being consumed) and as intermediates in the laboratory and in industry. The class includes such compounds as ferrocene, a remarkably stable compound in which an iron atom is sandwiched between two hydrocarbon rings.Organometallic compounds are typically discussed in terms of the metal as either main-group compounds or transition metal (transition element) compounds. The main-group metals of organometallic compounds are typically considered to be those of the S-block (groups 1 and 2) and the heavier elements of the p-block (groups 13–15) in the periodic table of elements. The transition metals include those elements in the d- and f-blocks (groups 3–12).
The physical and chemical properties of organometallic compounds vary greatly. Most are solids, particularly those whose hydrocarbon groups are ring-shaped or aromatic, but some are liquids and some are gases. Their heat and oxidation stability vary widely. Some are very stable, but a number of compounds of electropositive elements such as lithium, sodium, and aluminum are spontaneously flammable. Many organometallic compounds are highly toxic, especially those that are volatile.
The properties of the organometallic compounds depend in large measure on the type of carbon-metal bonds involved. Some are ordinary covalent bonds (covalent bond), in which pairs of electrons (electron) are shared between atoms. Others are multicentre covalent bonds, in which the bonding involves more than two atoms. A third type are ionic bonds (ionic bond), in which the bonding electron pair is donated by only one atom. In donor-acceptor bonds, the metal atom is connected to hydrocarbons with multiple bonds between carbon atoms.
Where metal atoms form covalent bonds with carbon atoms, the electrons are usually shared unequally. As a result, the bond is polarized—one end is more negative than the other. The extent of polarization depends on the strength with which the metal atom binds electrons. Organometallic compounds range in polar power from methylpotassium, in which the bond is almost like certain ionic bonds, to lead, which bonds with carbon with very little polarization.
Importance of organometallic compounds
Because of bond polarity, many organometallic compounds have reactivities that have made them important in chemical synthesis. The organomagnesium halides (Grignard reagents (Grignard reagent)), for example, are used widely in synthetic organic chemistry, as are organolithium and organoboron compounds. Alkylaluminum compounds are also employed in organic synthesis. Used with titanium salts, they are important catalysts in the polymerization of unsaturated hydrocarbons, such as ethylene and propylene. The mechanism of action of the titanium-aluminum alkyl catalysts probably involves interaction between the titanium atoms and the double bonds of the hydrocarbons.
Organometallic compounds containing lead, tin, and mercury are all commercially significant. A large number of organotin compounds, for example, are used as pharmaceuticals (pharmaceutical), pesticides (pesticide), stabilizers for polyvinyl chloride, and fire retardants. Methylmercury has caused severe pollution problems as a result of its toxicity. This fact has led to stringent controls on the discharge of mercury from chemical plants into rivers, lakes, and oceans.
carbon monoxide reacts readily with many transition-metal atoms to form metal carbonyls (metal carbonyl), themselves a class of organometallics. One of the earliest to be discovered was tetracarbonylnickel, a volatile nickel compound that became the basis of a process for purifying nickel. Metal carbonyls are employed as catalysts in many reactions in the petrochemical industry.
Defining characteristics
 A compound is regarded as organometallic if it contains at least one metal-carbon (M−C) bond where the carbon is part of an organic group. Typically, an organic group contains carbon-hydrogen (C−H) bonds; for example, the simple methyl group, CH3, and larger homologs such as the ethyl group, C2H5, which attach to a metal atom through only one carbon atom. (Simple alkyl groups such as these are often abbreviated by the symbol R.) More elaborate organic groups include the cyclopentadienyl group, C5H5, in which all five carbon atoms can form bonds with the metal atom. The term metallic is interpreted broadly in this context; thus, when organic groups are attached to the metalloids (metalloid) such as boron (B), silicon (Si), germanium (Ge), and arsenic (As), the resulting compounds are considered to be organometallic along with those containing true metals such as lithium (Li), magnesium (Mg), aluminum (Al), and iron (Fe). The “metal” in an organometallic compound can include most elements, with the exception of nitrogen (N) and phosphorus (P) in group 15 and all the elements in groups 16 (the oxygen group (oxygen group element)), 17 (halogens (halogen element)), and 18 (noble gases (noble gas)).
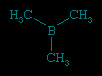
A compound is regarded as organometallic if it contains at least one metal-carbon (M−C) bond where the carbon is part of an organic group. Typically, an organic group contains carbon-hydrogen (C−H) bonds; for example, the simple methyl group, CH3, and larger homologs such as the ethyl group, C2H5, which attach to a metal atom through only one carbon atom. (Simple alkyl groups such as these are often abbreviated by the symbol R.) More elaborate organic groups include the cyclopentadienyl group, C5H5, in which all five carbon atoms can form bonds with the metal atom. The term metallic is interpreted broadly in this context; thus, when organic groups are attached to the metalloids (metalloid) such as boron (B), silicon (Si), germanium (Ge), and arsenic (As), the resulting compounds are considered to be organometallic along with those containing true metals such as lithium (Li), magnesium (Mg), aluminum (Al), and iron (Fe). The “metal” in an organometallic compound can include most elements, with the exception of nitrogen (N) and phosphorus (P) in group 15 and all the elements in groups 16 (the oxygen group (oxygen group element)), 17 (halogens (halogen element)), and 18 (noble gases (noble gas)).One example of an organometallic compound is trimethylboron, B(CH3)3, which contains three B−C bonds.
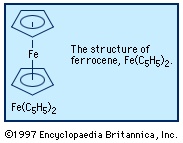 Another is ferrocene, Fe(C5H5)2, which has a more elaborate structure with the iron atom sandwiched between two C5H5 rings. Some compounds with metal-carbon bonds are not regarded as organometallic, because the constituent carbon atom is not part of an organic group; two examples are metal carbides (carbide)—such as Fe3C, a hard solid that is a component of cast iron—and metal cyanide compounds—such as the deep-blue paint pigment Prussian blue, KFe2(CN)6.
Another is ferrocene, Fe(C5H5)2, which has a more elaborate structure with the iron atom sandwiched between two C5H5 rings. Some compounds with metal-carbon bonds are not regarded as organometallic, because the constituent carbon atom is not part of an organic group; two examples are metal carbides (carbide)—such as Fe3C, a hard solid that is a component of cast iron—and metal cyanide compounds—such as the deep-blue paint pigment Prussian blue, KFe2(CN)6.Historical developments
 The first synthetic organometallic compound, K【PtCl3(C2H4)】, was prepared by the Danish pharmacist William C. Zeise in 1827 and is often referred to as Zeise's salt. At that time, Zeise had no way of determining the structure of his new compound, but today it is known that the structure contains an ethylene molecule (H2C=CH2) attached through both carbon atoms to the central platinum (Pt) atom. The platinum atom also is bonded to three chlorine (Cl) atoms. The potassium ion, K+, is present to balance the charge.
The first synthetic organometallic compound, K【PtCl3(C2H4)】, was prepared by the Danish pharmacist William C. Zeise in 1827 and is often referred to as Zeise's salt. At that time, Zeise had no way of determining the structure of his new compound, but today it is known that the structure contains an ethylene molecule (H2C=CH2) attached through both carbon atoms to the central platinum (Pt) atom. The platinum atom also is bonded to three chlorine (Cl) atoms. The potassium ion, K+, is present to balance the charge.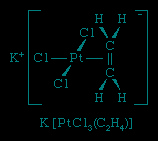
The attachment of the ethylene carbon atoms to the central platinum atom qualifies Zeise's salt as an organometallic compound. A development with a more immediate impact on the field of chemistry was the discovery in 1849 by the German-trained British chemist Edward C. Frankland (Frankland, Sir Edward) of diethylzinc, H5C2−Zn−C2H5, which he showed is very useful in organic synthesis. Since then, an ever-increasing variety of organometallic compounds have been utilized in organic synthesis in both the laboratory and industry.
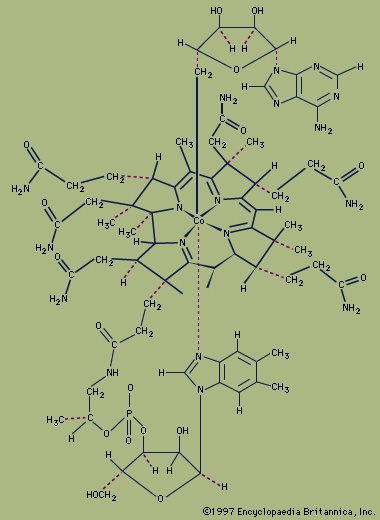 Another milestone in the development of the field was the discovery of tetracarbonylnickel by the German-educated British industrial chemist Ludwig Mond (Mond, Ludwig) and his assistants in 1890. In 1951, German theoretical chemist Ernst Otto Fischer (Fischer, Ernst Otto) and British chemist Sir Geoffrey Wilkinson (Wilkinson, Sir Geoffrey) independently discovered the sandwich structure of the compound ferrocene. Their parallel discoveries led to the subsequent unveiling of other compounds with sandwich structures, and in 1973, Fischer and Wilkinson were jointly awarded the Nobel Prize for Chemistry for their contributions to the study of organometallic compounds. Since the 1950s, organometallic chemistry has become a very active field, marked by the discovery of new organometallic compounds along with their detailed structural and chemical characterization and their application as synthetic intermediates and catalysts in industrial processes. Two organometallics encountered in nature are the coenzyme (vitamin B12), which contains a cobalt-carbon (Co−C) bond, and dimethylmercury, H3C−Hg−CH3, which is produced by bacteria to eliminate the toxic metal mercury. However, organometallic compounds are generally unusual in biological processes.
Another milestone in the development of the field was the discovery of tetracarbonylnickel by the German-educated British industrial chemist Ludwig Mond (Mond, Ludwig) and his assistants in 1890. In 1951, German theoretical chemist Ernst Otto Fischer (Fischer, Ernst Otto) and British chemist Sir Geoffrey Wilkinson (Wilkinson, Sir Geoffrey) independently discovered the sandwich structure of the compound ferrocene. Their parallel discoveries led to the subsequent unveiling of other compounds with sandwich structures, and in 1973, Fischer and Wilkinson were jointly awarded the Nobel Prize for Chemistry for their contributions to the study of organometallic compounds. Since the 1950s, organometallic chemistry has become a very active field, marked by the discovery of new organometallic compounds along with their detailed structural and chemical characterization and their application as synthetic intermediates and catalysts in industrial processes. Two organometallics encountered in nature are the coenzyme (vitamin B12), which contains a cobalt-carbon (Co−C) bond, and dimethylmercury, H3C−Hg−CH3, which is produced by bacteria to eliminate the toxic metal mercury. However, organometallic compounds are generally unusual in biological processes.s- and p-block organometallic compounds
The metal in main-group organometallic compounds can be any of the elements in the s block (i.e., groups 1 and 2) or any of the heavier elements in groups 13 through 15. (Groups 13–18 constitute the p block.) The elements at the borderline between the d block and p block—namely, zinc, cadmium, and mercury—will be discussed along with the p-block organometallics because of the similarity of their organometallic chemistry. In an internationally sanctioned system of nomenclature, the organic group is named first, followed by the metal, as in dimethylmercury. In writing the formula, this order is reversed, Hg(CH3)2. The organic groups, which are also called ligands (ligand), are named in the same way as for any organic compound. The number of carbon atoms on a group that are attached to the metal is indicated by the superscript in ηn. This convention is known as hapto nomenclature. A single point of attachment, η1, is usually not explicitly indicated, as in the above formula for dimethylmercury, a monohapto species. The compound with the common name ferrocene has the systematic name bis(η5-cyclopentadienyl)iron, where the number of cyclopentadienyl ligands (two) is indicated by the prefix bis and the number of sites of attachment (five) for each of these is indicated by η5. Ferrocene is thus called a pentahapto compound. The number of sites of attachment are also indicated in the formula Fe(η5-C5H5)2.The stability and reactivity (reactive dye) of organometallic compounds
The stability and reactivity of organometallic compounds are associated with the nature of the organic ligands and the metal to which they are attached. In each of the main groups of the periodic table (groups 1, 2, and 13–15), the thermal stability of a given type of organometallic compound generally decreases from the lightest to the heaviest element in a group. For example, in compounds containing group-1 metals, methyllithium (LiCH3) is much more stable than methylpotassium (KCH3), and, in those with group-14 metals, tetramethylsilicon, Si(CH3)4, is stable in the absence of air at 500 °C (932 °F), whereas tetramethyllead, Pb(CH3)4, rapidly decomposes at that temperature. This trend in stability is a consequence in part of the decrease in M−C bond strength on going down within a group. The trend does not hold for the d-block elements (groups 3–12), where M−C bond strengths and stability often increase going down a group.
The reactivities of organometallic compounds with water and air vary widely. The highly active main-group metals such as lithium (Li), sodium (Na), magnesium (Mg), and aluminum (Al) form highly air- and water-sensitive organometallic compounds. For example, Al2(CH3)6 undergoes immediate and violent reaction with water to liberate methane (CH4) gas, and it bursts immediately into flame on contact with air. For the elements toward the right of the periodic table (groups 14 and 15), the organometallic compounds are not water-sensitive; tetramethylsilicon, for example, does not react with water or air at room temperature.
The synthesis of s- and p-block organometallic compounds
Synthesis (chemical synthesis) of s- and p-block organometallic compounds can often be accomplished by one of several general reaction types. The most important of these are outlined below.
Formation of alkyllithium and Grignard reagents (Grignard reagent)
The highly active metals combine with a halogen (halogen element)-substituted hydrocarbon to produce simple organometallic compounds. For example, methyllithium, an important reagent in organic synthesis, is produced commercially by following the reaction:
2Li + CH3Cl → LiCH3 + LiCl
With other active metals, such as magnesium, aluminum, and zinc, the reaction generally yields the organometallic halide. A common reaction of this type is the synthesis of a Grignard reagent, an alkylmagnesium halide that finds wide use in organic synthesis (the s indicates that the metal is in solid form).
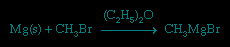
Double displacement
The synthesis of organometallic compounds by double displacement involves organometallic (MR) and binary halide (EX, where E is a metal or metalloid and X is a halogen) starting materials. It provides a convenient synthetic procedure that is widely used in the laboratory and to a lesser extent on a commercial scale. As the following examples illustrate, the organic group on the more active metal is transferred to the less active metal or metalloid. In this context the most common highly active metals are lithium, aluminum, and magnesium.
4Li(CH3) + SiCl4 → 4LiCl + Si(CH3)4
Al2(CH3)6 + 2BF3 → 2AlF3 + 2B(CH3)3
Redistribution
Double displacements involving the same central element are often referred to as redistribution reactions. A commercially important example is the redistribution of silicon tetrachloride and tetramethylsilicon (also known as tetramethylsilane) at elevated temperatures.
SiCl4 + (CH3)4Si → CH3SiCl + (CH3)2SiCl2 + (CH3)3SiH + ...
The products from this reaction can be separated by distillation. This reaction is performed industrially where (CH3)2SiCl2 is removed from the equilibrating mixture and then hydrolyzed to produce the intermediates for silicone polymers, which have the form −(Si(CH3)2−O)−n (For more information about the properties and synthesis of inorganic polymers, see inorganic polymer (polymer, inorganic)).
Hydrometallation
The addition of a metal hydride to a multiple bond is called hydrometallation, and it leads to the formation of a metal-carbon bond.
M−H + H2C=CH2 → MH2C−CH3
This reaction is driven mainly by the high C−H bond strength relative to most E−H bond strengths. Two important hydrometallation reactions are hydroboration and hydrosilation, illustrated, respectively, by the following examples.
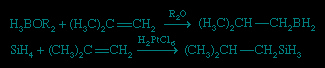
In the hydroboration and hydrosilation of an unsymmetrical alkene (olefin), the boron or silicon binds to the carbon atom that has less-bulky substituents, and the smaller hydrogen atom binds to the carbon atom that has bulky substituents—(CH3)2C in the above equations. Hydroboration was discovered and developed in the United States by Herbert C. Brown (Brown, Herbert Charles), who shared the Nobel Prize for Chemistry in 1979 for this research. Both hydroboration and hydrosilation are widely used in the synthesis of complex organic molecules. In these applications, the B−C or Si−C bond is generally cleaved in a subsequent step to produce a product that is free of boron or silicon.
reduction
All organometallic compounds are potential reducing agents, and those of the electropositive elements are very strong reducing agents because the metal gives up electrons (electron) to the carbon, resulting in a polar M−C bond with a partial positive charge on the metal and a negative charge on the carbon. Organometallic compounds of highly electropositive elements such as lithium, sodium, and aluminum ignite spontaneously and sometimes explode on contact with air or other oxidizing agents. The useful organometallic reagents Li(CH3), Zn(CH3)2, B(CH3)3, and Al2(CH3)6 are spontaneously flammable in air (pyrophoric). Accordingly, techniques have been developed to handle these and other pyrophoric compounds. glass reaction vessels sealed from the atmosphere and purged with nitrogen gas are commonly used for handling air-sensitive organometallic compounds in the laboratory. Large quantities of pyrophoric compounds such as Al2(C2H5)6 are routinely handled with ease in the chemical industry by using closed metal reactors (reactor) for the production of these and other much less reactive compounds. Organometallic compounds have reduced reactivity when the metallic component is not highly electropositive and when the metal is completely surrounded by attached groups. For example, elevated temperatures are required to initiate combustion with Si(CH3)4 and Sn(CH3)4, and at room temperature they can be handled in air.
Carbanion character
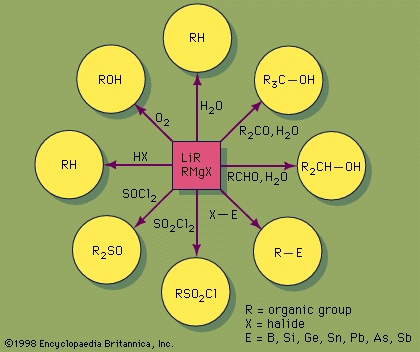 The partial negative charge of an organic group bonded to a highly active metal results in a distinctive pattern of reactivity that is frequently referred to as nucleophilic or carbanion character. Thus, organometallic compounds containing highly active (electropositive) metals, such as lithium, magnesium, aluminum, and zinc, react rapidly and completely with water, liberating a hydrocarbon in the process. For example, dimethylzinc liberates methane gas along with solid zinc hydroxide.
The partial negative charge of an organic group bonded to a highly active metal results in a distinctive pattern of reactivity that is frequently referred to as nucleophilic or carbanion character. Thus, organometallic compounds containing highly active (electropositive) metals, such as lithium, magnesium, aluminum, and zinc, react rapidly and completely with water, liberating a hydrocarbon in the process. For example, dimethylzinc liberates methane gas along with solid zinc hydroxide.Zn(CH3)2 + 2H2O → Zn(OH)2 + 2CH4
The above hydrolysis of dimethylzinc can be viewed as a transfer of a slightly acidic H+ from water to the strongly basic carbanion CH3− in dimethylzinc.
Alkyllithium, alkylaluminum, and alkylmagnesium compounds are the most common carbanion reagents in laboratory-scale synthetic chemistry; carbanion character is greatly diminished for the less metallic elements boron and silicon. The nucleophilic character of organometallic compounds of active metals has many synthetic applications. For example, the organic group in organometallic compounds of active metals attacks the carbonyl carbon of a ketone, and upon hydrolysis a tertiary alcohol results. Similarly, aldehydes (aldehyde) can be converted to secondary alcohols by reaction with an organometallic reagent followed by hydrolysis. Double displacement reactions can be used to prepare sulfones (sulfone) (R2SO2) and sulfoxides (sulfoxide) (R2SO) by treating thionyl chloride (SOCl2) or sulfuryl dichloride (SO2Cl2) with an alkyllithium or a Grignard reagent.
One consequence of the carbanion character of organometallic compounds containing active metals is the protolysis ( proton-transfer) reaction that takes place with very weak protonic acids, including water. Alcohols react in a manner similar to the reaction of water, and this provides a convenient way of introducing an alkoxide (OR) substituent into an organometallic compound.
(C2H5)3Ga + HOCH3 → 【(C2H5)3GaOHCH3】 → (C2H5)2Ga(OCH3) + C2H6
The rate of reaction decreases with bulky organic groups on the alcohol. For example, tert-butyl alcohol,
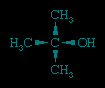
reacts slowly with most active organometallics, and it is therefore employed in the laboratory to safely destroy reactive organometallic wastes.
β-hydrogen elimination
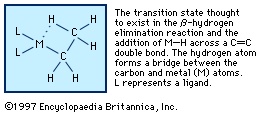 This is a common organometallic reaction in which a hydrogen atom on a carbon atom that is one position removed from the metal (the β position) transfers to the metal with the liberation of an alkene (olefin). The following example shows the formation of ethylene, C2H4.
This is a common organometallic reaction in which a hydrogen atom on a carbon atom that is one position removed from the metal (the β position) transfers to the metal with the liberation of an alkene (olefin). The following example shows the formation of ethylene, C2H4.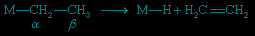
This reaction is the reverse of the addition of an M−H bond to an alkene (see above Hydrometallation (organometallic compound)), and under some conditions significant equilibrium concentrations of both reactants and products are observed. The β-hydrogen elimination reaction is thought to proceed through the transition state, and as might be expected from this reaction mechanism, compounds with an accessible central metal atom tend to undergo β-hydrogen elimination readily. For example, it occurs with trialkylaluminum compounds but not with tetraalkylsilicon compounds, in which reaction of the silicon atom is hindered by the bulky substituents.
d- and f-block organometallic compounds
The nomenclature for organometallic compounds of the d-block elements, also known as transition metals (transition element), follows rules similar to those described for the main-group organometallics. Groups attached to the metal (ligands) are named first, followed by the metal. When a ligand is attached to the metal by more than one carbon atom, the number of sites of attachment is indicated by n in ηn.
Metal carbonyls (metal carbonyl)
Many of the early discoveries in d-block organometallic chemistry involved the metal carbonyls (carbonyl group)—i.e., compounds consisting of a metal atom bonded to one or more carbon monoxide (CO) ligands. Mond (Mond, Ludwig)'s discovery of the first simple metal carbonyl, tetracarbonylnickel, Ni(CO)4, at the end of the 19th century was quickly followed by a series of discoveries in his laboratory and elsewhere showing that most of the d-block metals form neutral homoleptic carbonyls. (The term homoleptic refers to identical groups attached to a central atom.) The remarkable volatility of tetracarbonylnickel, whose boiling point is 43 °C, prompted one of Mond's contemporaries to state that “Mond put wings on metals.” The ease of formation of tetracarbonylnickel (from metallic nickel and carbon monoxide) and its high volatility led Mond to develop an efficient industrial process for separating nickel and cobalt, which often occur together in their ores (ore). This process requires diligent safety measures because tetracarbonylnickel is extremely toxic.
The structure of metal carbonyls
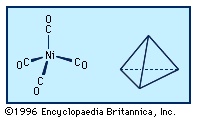
The carbonyl ligands in the tetracarbonylnickel molecule project toward the vertices of a tetrahedron, and thus the structure is referred to as tetrahedral. Similarly, the six carbonyl ligands in hexacarbonylchromium project toward the vertices of an octahedron.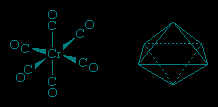
Many other metal carbonyls contain two or more metal atoms, such as decacarbonyldimanganese and octacarbonyldicobalt, shown here.
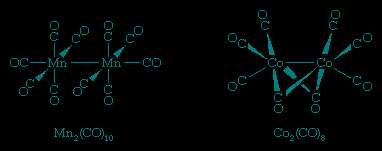
In all these structures carbon monoxide is connected to the metal through its carbon atom. When more than one metal atom is present, as in octacarbonyldicobalt, the carbon of the carbonyl ligand may bridge between metal atoms.
The formulas of most homoleptic metal carbonyl compounds conform to the rule that each metal atom in the metal carbonyl molecule must have 18 valence electrons (valence electron). The valence electrons represent the outer electrons on the metal plus those from the ligand. For example, the electron count for Fe(CO)5 includes eight from the iron atom (it is in group 8 and hence has eight valence electrons) and two from each carbonyl ligand, giving a total of 18 electrons. This 18-electron rule applies to many organometallic compounds of the d-block metals other than carbonyls, but there are exceptions in the organometallic chemistry of the d-block metals. The most notable exceptions are metals on the far left of the d block (e.g., V(CO)6 contains 17 electrons) and on the right of the d block where the platinum metals (platinum group)— rhodium (Rh), iridium (Ir), palladium (Pd), and platinum (Pt)—often exhibit a 16-electron count in their organometallic compounds.
Zero-oxidation-state metal carbonyls
 The central metal in a neutral metal carbonyl, such as those described above, is assigned an oxidation state of zero, quite unlike the case in simple inorganic compounds in which positive oxidation states are the norm, as, for example, Fe3+ in FeCl3 or Ni2+ in NiBr2. Unlike the free metals, which also have a zero oxidation state, many carbonyls are soluble in a variety of simple organic solvents and are highly reactive. Because of these chemical and physical properties, the metal carbonyls are convenient starting materials for the synthesis of compounds with the metal atom in a zero or low oxidation state. One simple reaction is the substitution of other ligands such as triethylphosphine, P(Et)3, for CO (Et is a common abbreviation for the ethyl group, −C2H5).
The central metal in a neutral metal carbonyl, such as those described above, is assigned an oxidation state of zero, quite unlike the case in simple inorganic compounds in which positive oxidation states are the norm, as, for example, Fe3+ in FeCl3 or Ni2+ in NiBr2. Unlike the free metals, which also have a zero oxidation state, many carbonyls are soluble in a variety of simple organic solvents and are highly reactive. Because of these chemical and physical properties, the metal carbonyls are convenient starting materials for the synthesis of compounds with the metal atom in a zero or low oxidation state. One simple reaction is the substitution of other ligands such as triethylphosphine, P(Et)3, for CO (Et is a common abbreviation for the ethyl group, −C2H5).Cr(CO)6 + P(Et)3 → Cr(CO)5P(Et)3 + CO
Because most other ligands are far bulkier than carbon monoxide and also because of differences in bonding properties, it is often not possible to replace all the carbonyl ligands by groups such as triethylphosphine, and a mixture of products with varying degrees of substitution can result.
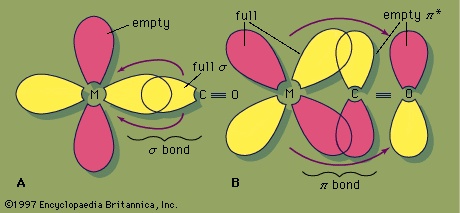
The remarkable ability of the carbonyl ligand to stabilize compounds with metals in the zero oxidation state has led to detailed studies of M−CO bonds. A minor part of the M−CO bonding can be attributed to the tendency of the carbonyl ligand to donate a pair of electrons to the metal, which is the principal mode of interaction for most ligands with metal cations (cation) in classical coordination compounds (coordination compound) such as 【Co(NH3)6】3+. The second mode of interaction with the metal is the simultaneous back-donation of electron density from the metal to the carbonyl ligand, which is called back π bonding.
Metal carbonyl anions
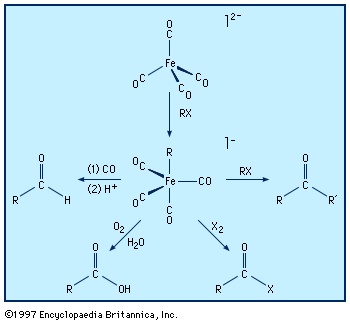 More remarkable than the formation of zero-oxidation-state metal carbonyls (carbonyl group) is the reduction of many of these carbonyl compounds to metal carbonyl anions (anion), in which the metal has a negative oxidation state. The following example demonstrates that the two- electron reduction by sodium metal is accompanied by the loss of the two-electron donor carbonyl ligand, and so the 18-electron count on iron is preserved; the solvent is tetrahydrofuran (THF).
More remarkable than the formation of zero-oxidation-state metal carbonyls (carbonyl group) is the reduction of many of these carbonyl compounds to metal carbonyl anions (anion), in which the metal has a negative oxidation state. The following example demonstrates that the two- electron reduction by sodium metal is accompanied by the loss of the two-electron donor carbonyl ligand, and so the 18-electron count on iron is preserved; the solvent is tetrahydrofuran (THF).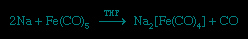
The stabilization of very electron-rich complexes, such as 【Fe(CO)4】2−, is attributed to the back π bonding that shifts electron density from the metal to the carbonyl ligands, and this view is substantiated by C−O bond distances and other physical data. The metal atom in these carbonyl anions is assigned a negative oxidation state (−2 in the above example). This formalism does not acknowledge the delocalization of electron density from the metal to the ligand, but the chemical properties of the carbonyl anions do suggest that some of the negative charge resides on the metal. For example, a metal carbonyl anion can be protonated with the H+ ion, which generally attaches to the central metal and not a carbonyl ligand, as in the following example.
【Fe(CO)4】2− + HCl → 【HFe(CO)4】− + Cl−
Owing to their high reactivity, carbonyl anions are useful starting materials for the synthesis of other organometallic compounds, and this accounts for their applications in organic synthesis. For example, 【Fe(CO)4】2− is used to extend a carbon chain by transfer of the carbonyl substituent, producing aldehydes (aldehyde), ketones (ketone), or carboxylic acids (carboxylic acid).
Compounds with metal-carbon bonds
The variety of hydrocarbon ligands found in d-block organometallic chemistry range from simple σ-bonded alkyl ligands, double-bonded carbenes (carbene), and triply bonded carbynes to a host of polyene ligands, some of which are described in the remainder of this section.
Simple alkyl ligands
A simple alkyl ligand forms an M−C single bond, and in doing so the alkyl group acts as a one-electron monohapto ligand. Many such compounds are known, but they are less common in the d block than in the s and p blocks. This may in part be a result of the modest M−C bond strengths. Another reason for the limited stability of alkyl ligands in many d-block complexes is a set of reactions that can be quite rapid, such as β-hydrogen elimination, CO insertion, and reductive elimination. As described below, these reactions transform simple hydrocarbon ligands into other groups.The β-hydrogen elimination reaction is an important feature of the hydrocarbon chemistry of both d- and p-block organometallics. The reaction consists of the abstraction of a hydrogen atom from the organic ligand with the formation of two products, one of which contains a metal-hydrogen bond and the other of which is an alkene.
LnM−CH2CH3 → LnM−H + H2C=CH2
(Ln represents the n ligands not involved in the hydrogen elimination.) The reverse of this reaction, alkene insertion into the M−H bond, is illustrated by the hydroboration and hydrosilation reactions discussed above. Both the β-hydrogen elimination and addition of M−H across a C=C double bond are thought to proceed through a cyclic intermediate involving a three-centre, two-electron bond where a hydrogen atom bridges between the carbon and the metal atoms.
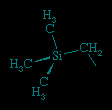
Since a compound with no hydrogen atoms on the β-carbon atom cannot undergo β-hydrogen elimination, benzyl, CH2(C6H5), and methyltrimethylsilyl, CH2Si(CH3)3 (shown below) ligands are generally more robust than ethyl ligands when attached to d-block metal atoms. Similarly, the lack of β-hydrogen atoms on the methyl group accounts for the greater stability of complexes containing the methyl ligand rather than the ethyl ligand.
A reaction frequently referred to as CO insertion leads to carbon-carbon bond formation between the carbon atom of a carbonyl ligand and the carbon atom of an alkyl ligand, which is the methyl group in the following example.

The CO insertion reaction is involved in all the transformations of 【Fe(CO)4R】− into organic molecules.
Another type of reaction that can transform an attached organic ligand (as well as other groups) is reductive elimination.
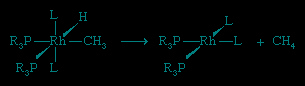
The converse of reductive elimination is oxidative addition.
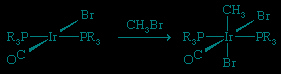
The reactions discussed above, insertion of C=C into an M−H bond, β-hydrogen elimination, and CO insertion, are often employed for the synthesis of organic molecules in the laboratory and in industry. They also account for some of the individual steps in some important catalytic cycles (see below Organometallic compounds in catalysis (organometallic compound)).
Alkylidene ligands
Alkylidene ligands, such as CH2, CHR, or CR2, form the M=C d-p double bonds (i.e., bonds between the d orbitals (orbital) of the metal and the p orbitals of the carbon), and their metal compounds are often called carbenes. The first stable metal carbene complex was discovered in the laboratory of the German chemist Ernst O. Fischer (Fischer, Ernst Otto) in Munich by the reaction of a metal carbonyl with an alkyllithium compound. In this reaction, the alkyl group is transferred (as an R− group) to the carbon atom of the coordinated carbonyl, and subsequent addition of carbocation reagent results in attachment of an R+ group to the oxygen.
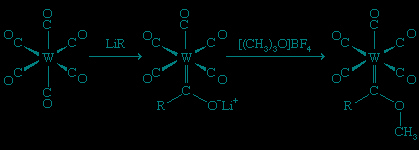
This type of carbene complex is common for the atoms of metals in groups 6–8, and they are called Fischer carbenes. The Fischer carbenes can be modified by electron-rich groups. For example, the attack of an amine on the electron-poor carbon atom of a Fischer carbene results in the displacement of the OR group to yield a new carbene (Me represents the methyl group, −CH3).

The attack of an electron-rich amine (indicated by δ- in the above equation) on the carbon atom in the Fischer carbene is attributed to the significant electronegativity of the middle to late d-block metals, which makes the carbon atom of the carbene electron-poor (indicated by δ+ in the above equation).
The reactions of Fischer carbene complexes with alkynes has considerable utility in organic synthesis. For example, naphthyl compounds (i.e., those derived from the fused ring system C10H8) can be synthesized by the reaction of methoxy phenyl Fischer carbenes with an alkyne.
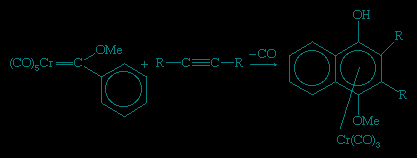
The chromium can be removed from the organic product by mild oxidation. Another route to a carbene is the deprotonation of an alkyl ligand, producing a carbene that contains only hydrocarbon ligands.
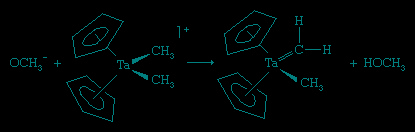
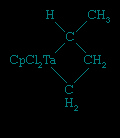
These ligands that contain only carbon and hydrogen are commonly attached to metal atoms from the early part of the d block such as titanium (Ti) and tantalum (Ta). The complexes are known as Schrock carbenes for their discoverer, American chemist Richard Schrock (Schrock, Richard R.). The chemistry and spectroscopy of the Schrock carbenes indicate that these compounds have the opposite polarity of the Fischer carbenes. The carbon behaves as if it were electron-rich, because the Mδ+=Cδ− bond is polarized so as to put negative charge on the metal-bound carbon, for the early d-block metals readily give up electrons. As a result, the carbon attached to the metal atom in a Schrock carbene reacts with electron-seeking reagents, such as Me3Siδ+Brδ+, at the carbene carbon (Cp is a common abbreviation for the cyclopentadienyl ligand, −C5H5).
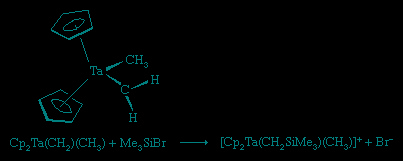
An interesting reaction of the Schrock carbenes is the alkene metathesis reaction:
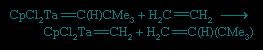
This reaction appears to proceed through a four-membered ring intermediate containing carbon atoms and the metal atom.
Alkylidyne ligands
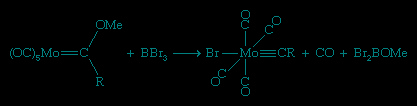
Alkylidyne ligands have the general formula CH or CR. They are bound to the metal by an M≡C triple bond involving one σ bond and two d-p π bonds. The simplest member of this series is methylidyne, CH, and the next simplest, CCH3, is ethylidyne. One route to methylidynes, discovered in Fischer's laboratory, involves the abstraction of an alkoxide group (OR) from a Fischer carbene by BBr3.
Alkene (olefin) and alkyne ligands
An alkene (olefin) ligand contains a π bond between carbon atoms, C=C, which can serve as an electron pair donor in a metal complex, as in the case of Zeise's salt (see above Historical developments (organometallic compound)). This complex may be prepared by bubbling ethylene, C2H4, through an aqueous solution of 【PtCl4】2− in the presence of divalent tin, Sn(II), which aids in the removal of the chloride ion (Cl−) from the coordination sphere of the divalent platinum, Pt(II).

The alkene ligand bonds to the metal centre by both electron donation and acceptance, similar to the situation with carbon monoxide. Electron donor-and-acceptor character between the metal and the alkene ligand appear to be fairly evenly balanced in most ethylene complexes of the d metals.
The allyl ligand, −CH2−CH=CH2, can bind to a metal atom in either of two configurations: as an η1-ligand or an η3-ligand. Because of this versatility in bonding, η3-allyl complexes are often highly reactive. Examples of η1- and η3-allyl complexes are, respectively, shown here.
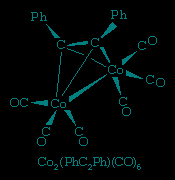
acetylene, H−C≡C−H, has two π bonds and hence is a potential four-electron donor. Substituted acetylenes form very stable polymetallic complexes in which the acetylene can be regarded as a four-electron donor. An example is η2-diphenylethynehexacarbonyldicobalt, in which four of the six electrons in the triple bond of the ethyene ligand, R−C≡C−R, are shared with the two cobalt atoms (Ph represents the phenyl ligand, −C6H5). As in this example, the alkyl or aryl groups (R) on the acetylene impart stability to the metal complex—in contrast to simple acetylene (HC≡CH) complexes, where the hydrogen atoms are reactive.
Polyene ligands
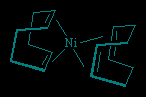
Diene (−C=C−C=C−) and larger polyene ligands present the possibility of several points of attachment to a metal atom. The resulting polyene complexes are usually more stable than the equivalent monohapto complex with individual ligands. For example, bis(η4-cycloocta-1,5-diene)nickel is more stable than the corresponding complex containing four ethylene ligands.
Cycloocta-1,5-diene (cod), a fairly common ligand in organometallic chemistry, is introduced into the metal coordination sphere by ligand displacement reactions; for example,
PdCl2(NCPh)2 + cod → codPdCl2 + 2NCPh.
Metal complexes of cod are often used as starting materials because the cod ligand can bind in various ways to the metal and the complexes are intermediate in stability. Many of them are sufficiently stable to be isolated and handled, but cod and similar ligands can be displaced by stronger ligands. For example, Ni(cod)2 reacts with CO to form Ni(CO)4 and the free cod molecule. This reaction is a convenient source of the extremely toxic Ni(CO)4, for it can be generated directly in a flask where it is then available to undergo a subsequent reaction.
Cyclic polyene ligands
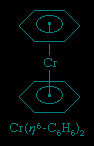
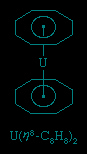
These rings, which have alternating double and single bonds, are among the most important ligands in organometallic chemistry; the most common members of this group range from cyclobutadiene (C4H4) to cyclooctatetraene (C8H8). Their organometallic compounds include the metallocenes ferrocene and bisbenzenechromium and bis(cyclooctatrienyl)uranium (commonly called uranocene), shown here.
A metallocene consists of a metal atom between two planar polyhapto rings (as in ferrocene), and because of this structure they are informally called sandwich compounds. Cyclic polyenes are also known to form complexes in which they bind to a metal atom through some but not all of their carbon atoms.
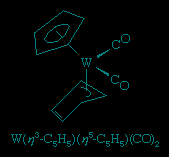
The cyclobutadiene ligand is a four-electron donor. It is unstable as the free (i.e., uncombined) hydrocarbon, but it is known to exist in stable complexes, including Ru(C4H4)(CO)3.
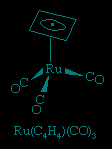
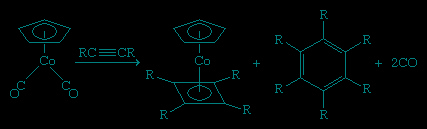
This is one of many cases in which coordination to a metal atom stabilizes an otherwise unstable molecule. Because of its instability, cyclobutadiene must be generated in the presence of the metal to which it is to be coordinated. This can be accomplished in several ways, one of which is the dimerization of a substituted acetylene. Interestingly, the C4R4 is bound to the cobalt in preference to the trimerization product, C6R6.
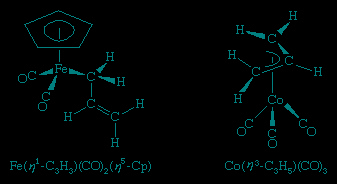
The cyclopentadienyl ligand (C5H5, abbreviated Cp) has played a major role in the development of organometallic chemistry. In some metal cyclopentadienyl compounds, the metal is bonded to only one of the five carbon atoms, and in these complexes the Cp is designated as a monohapto, η1-, ligand, which contributes one electron to form a σ bond with the metal, as in
Others contain a trihapto (η3-) cyclopentadienyl ligand, which donates three electrons. The most common case, however, is when Cp is a pentahapto ligand contributing five electrons. Two of the bonding modes for Cp are illustrated in the following structure, which contains both η3- and η5-C5H5 ligands.
The bis(η5-cyclopentadienyl)-sandwich complexes of iron, cobalt, and nickel are readily prepared by the reaction of sodium cyclopentadienide with the corresponding d-metal halide.

Because of their great stabilities, the 18-electron group-8 compounds ferrocene, ruthenocene, and osmocene maintain their ligand-metal bonds under rather harsh conditions, and it is possible to carry out a variety of reactions on the cyclopentadienyl ligands while they are attached to the central metal. In some cases, they undergo reactions similar to those of simple aromatic hydrocarbons, such as Friedel-Crafts substitution, which is a characteristic reaction of benzene, C6H6.
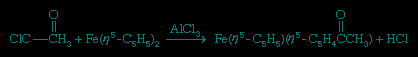
It is also possible to replace the hydrogen atom on a C5H5 ring with a lithium atom using the highly reactive reagent butyllithium.
LiC4H9 + Fe(η5-C5H5)2 → Fe(η5-C5H5)(η5-C5H4Li) + C4H10
This lithiated product is an excellent starting material for the synthesis of other ring-substituted products.
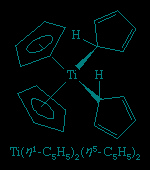
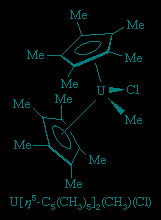
A closely related set of so-called bent sandwich compounds, in which the Cp rings are not parallel, are important in the organometallic chemistry of the early and middle d-block elements and the f-block elements (lanthanides and actinides (actinide element)). The Schrock carbene Ta(η5-C5H5)2(CH3)(CH2), shown above, is one such example. Bent sandwich compounds are important in the organometallic chemistry of the f-block elements, but to achieve stability the pentamethylcyclopentadienyl ligand, C5(CH3)5, is generally employed with these elements, as, for example, in the following uranium compound.
Metal clusters
Metal cluster compounds contain metal-metal bonds. The focus here is on compounds having three or more metals in a closed array. carbon monoxide is the most common ligand in organometallic cluster compounds, but many other organometallic ligands are bound to clusters, and the presence of several metals leads to bonding arrangements for the ligand that are not possible for monometallic compounds. A variety of metal arrays are seen in cluster compounds. Triangular, tetrahedral, and octahedral clusters are common, and much larger metal arrays are known. The structures of many clusters, which can be precisely determined by single-crystal X-ray diffraction, provide some clues to the way in which ligands are bound to the surfaces of bulk metal particles. The latter are more difficult to structurally characterize than are molecular clusters.
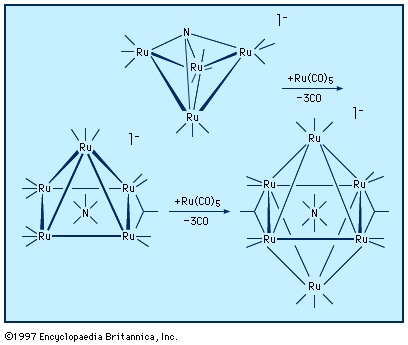 For many d-block clusters there is a strong correlation between their structure and the number of valence electrons (valence electron) (from the metal atoms and the ligands). This set of correlations for clusters is similar to the 18-electron rule for mononuclear organometallics, and these guidelines are often called Wade's rules after the British chemist Kenneth Wade, who first recognized that a triangular cluster such as Ru3(CO)12 usually has 48 valence electrons, a tetrahedron such as Co4(CO)12 has 60 electrons, and an octahedron such as Rh6(CO)12(μ3-CO)4 has 86 electrons. In some cases, it is possible to synthesize clusters in a stepwise manner. An interesting example of this type is the buildup of a ruthenium nitride cluster; in the process of cluster building, the nitrogen ligand is progressively encapsulated by metal atoms.
For many d-block clusters there is a strong correlation between their structure and the number of valence electrons (valence electron) (from the metal atoms and the ligands). This set of correlations for clusters is similar to the 18-electron rule for mononuclear organometallics, and these guidelines are often called Wade's rules after the British chemist Kenneth Wade, who first recognized that a triangular cluster such as Ru3(CO)12 usually has 48 valence electrons, a tetrahedron such as Co4(CO)12 has 60 electrons, and an octahedron such as Rh6(CO)12(μ3-CO)4 has 86 electrons. In some cases, it is possible to synthesize clusters in a stepwise manner. An interesting example of this type is the buildup of a ruthenium nitride cluster; in the process of cluster building, the nitrogen ligand is progressively encapsulated by metal atoms.Organometallic compounds in catalysis
Catalysts (catalyst) are substances that increase the rate of a reaction but are not consumed in the reaction. Catalysts are widely encountered in nature, industry, and the laboratory. Many of the catalysts utilized in the chemical industry and the laboratory are organometallic compounds.
hydrogenation
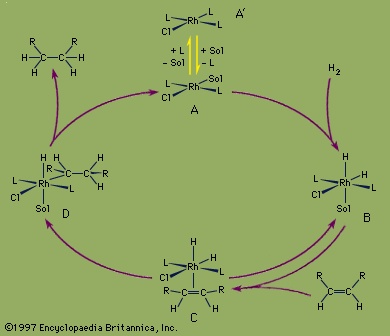 The overall result of the catalytic hydrogenation of alkenes (olefin) is to add molecular hydrogen, H2, across the double bond of an alkene. The reactants, H2 and ethylene (C2H4), enter the cycle by reaction with the complex to produce in succession a hydrido complex and an alkene complex. In the final step, the hydrogenated product leaves the loop with the regeneration of the coordinatively unsaturated Rh complex. The cycle continues as long as hydrogen gas and ethylene are supplied. The rhodium complexes in solution are the catalysts and are not used up in the reaction. Rhodium, which is more expensive than platinum, can be used even in catalytic processes where the products are inexpensive, because the rhodium is not consumed. Modifications of this type of catalyst are also employed in the production of pharmaceuticals such as levodopa (or L-dopa), which is used to treat Parkinson disease.
The overall result of the catalytic hydrogenation of alkenes (olefin) is to add molecular hydrogen, H2, across the double bond of an alkene. The reactants, H2 and ethylene (C2H4), enter the cycle by reaction with the complex to produce in succession a hydrido complex and an alkene complex. In the final step, the hydrogenated product leaves the loop with the regeneration of the coordinatively unsaturated Rh complex. The cycle continues as long as hydrogen gas and ethylene are supplied. The rhodium complexes in solution are the catalysts and are not used up in the reaction. Rhodium, which is more expensive than platinum, can be used even in catalytic processes where the products are inexpensive, because the rhodium is not consumed. Modifications of this type of catalyst are also employed in the production of pharmaceuticals such as levodopa (or L-dopa), which is used to treat Parkinson disease.Hydroformylation
Hydroformylation involves the addition of carbon monoxide and hydrogen to an alkene to form an aldehyde containing one more carbon atom than the original alkene.
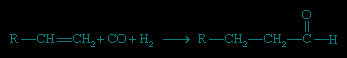
This catalytic reaction is employed in the petrochemical industry, where Co2(CO)8 or various rhodium catalysts are utilized. The catalytic cycle proceeds through a series of organometallic intermediates. The aldehydes produced by hydroformylation are normally reduced to alcohols (alcohol) that are used as solvents, as plasticizers, and in the synthesis of detergents. The scale of production is enormous, amounting to millions of tons per year.
Alkene polymerization
Polyalkenes (polyolefin), the most common and useful class of synthetic polymers (polymer), are often prepared by use of organometallic catalysts, either in solution or supported on a solid surface. In the 1950s, the German chemist Karl Ziegler (Ziegler, Karl) developed a catalyst for ethylene polymerization based on a catalyst formed by the reaction of TiCl4 with Al(C2H5)3. Soon thereafter, Italian chemist Giulio Natta (Natta, Giulio) made use of this type of catalyst for the polymerization of propylene to produce polymers with highly regular structures. The intimate details of the reactions of these commercial catalytic processes are not entirely understood, but there are strong indications from more easily studied soluble organometallic catalysts that alkenes coordinate to a metal centre and then insert into a hydrocarbon chain, producing a longer-chain hydrocarbon attached to the metal centre. Repetition of this process leads to extremely-long-chain hydrocarbon polymers, which include many of the most familiar plastics (plastic), such as polyethylene and polypropylene. These plastics are used in consumer items ranging from milk containers and plastic bags to artificial limbs and car bumpers.
Additional Reading
Introductory works
Introductory surveys of organometallic chemistry may be found in Duward F. Shriver and Peter Atkins, Inorganic Chemistry, 4th ed. (2005); and Gary L. Miessler and Donald A. Tarr, Inorganic Chemistry, 3rd ed. (2004).
Detailed works on structures and applications
Specific aspects of organometallic compounds are explored in Christoph Elschenbroich, Organometallics, 3rd ed. (2006), a well-illustrated discussion of s-, p-, and d-block organometallics, with emphasis on organometallic reactions; Akio Yamamoto, Organotransition Metal Chemistry, trans. from Japanese (1986), a very good presentation of the fundamentals and their application of d-block organometallic chemistry to synthesis and catalysis; Robert H. Crabtree, The Organometallic Chemistry of the Transition Metals, 3rd ed. (2000), stressing concepts of structure and reactions for d-block organometallics; James P. Collman et al., Principles and Applications of Organotransition Metal Chemistry, new ed. (1987), a highly authoritative and detailed presentation of d-block organometallic chemistry, with emphasis on principles, mechanisms, and the utility of organometallics in organic synthesis and catalysis; D. Michael P. Mingos and David J. Wales, Introduction to Cluster Chemistry (1990), emphasizing structure and bonding in d-block organometallic clusters; Duward F. Shriver, Herbert D. Kaesz, and Richard D. Adams (eds.), The Chemistry of Metal Cluster Complexes (1990), covering the structures and chemistry of d-block organometallic clusters, by authorities in the field; and George W. Parshall and Stephen D. Ittel, Homogeneous Catalysis, 2nd ed. (1992), treating commercial and laboratory homogeneous catalytic processes, most involving organometallic chemistry.
- Sir Desmond MacCarthy
- Sir Dominic John, Baronet Corrigan
- Sir Dominic John Corrigan, Baronet
- Sir Donald Coleman Bailey
- Sir Donald Currie
- Sir Donald Francis Tovey
- Sir Douglas Mawson
- Sir Dudley North
- Sir Dugald Clerk
- Sir Earle Page
- Sir Ebenezer Howard
- sire de Beaumanoir, Philippe de Remi
- sire de Joinville, Jean
- Sir Edmund Andros
- Sir Edmund Barton
- Sir Edmund Godfrey
- Sir Edmund Gosse
- Sir Edmund Hillary
- Sir Edmund Taylor Whittaker
- Sir Edward, 2nd Baronet Petre
- Sir Edward, 3rd Baronet Grey
- Sir Edward Albert Sharpey-Schafer
- Sir Edward Belcher
- Sir Edward Bullard
- Sir Edward Burnett Tylor