价键理论(卷名:化学)
valence-bond theory
一种获得分子薛定谔方程近似解的处理方法,也称电子配对法。是历史上最早发展起来的化学键理论,它主要描述分子中的共价键和共价结合,其核心思想是电子配对形成定域化学键。
简史 1927年德国物理学家W.H.海特勒和F.W.伦敦首次完成了氢分子中电子对键的量子力学近似处理,这是近代价键理论的基础。美国理论化学家J.C.斯莱特和L.C.鲍林把它推广应用于较复杂的分子。这个理论的特点在于强调分子中电子的定域性质,共价键是由原子的价电子交换而形成。
海特勒-伦敦方法处理氢分子 氢分子的哈密顿算符是:
 式中rA1、rB1为核A、B与电子1之间的距离;r12为两个电子之间的距离;RAB为两个原子核之间的距离……(图1);
式中rA1、rB1为核A、B与电子1之间的距离;r12为两个电子之间的距离;RAB为两个原子核之间的距离……(图1);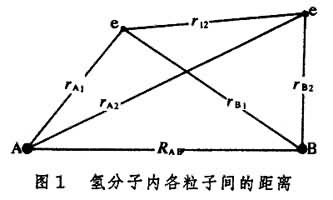 1/RAB表示两个原子核之间的势能(氢核和电子电荷皆为 1基本电荷单位);1/rA1、1/rB1、…也是势能;墷2是拉普拉斯算符。
1/RAB表示两个原子核之间的势能(氢核和电子电荷皆为 1基本电荷单位);1/rA1、1/rB1、…也是势能;墷2是拉普拉斯算符。海特勒-伦敦方法的要点在于如何恰当地选取基态H2的近似波函数Ψ(1,2)(或称尝试波函数),然后用变分公式使氢分子能量 E为最低(假定Ψ是归一化的):
 式中*表示复数共轭。考虑两个氢原子组成的体系,若两个氢原子A(有电子1)和B(有电子2)的基态波函数为:
式中*表示复数共轭。考虑两个氢原子组成的体系,若两个氢原子A(有电子1)和B(有电子2)的基态波函数为:φA(1)=π-1/2exp(-rA1)
φB(2)=π-1/2exp(-rB2)假如两个氢原子相距很远,那么体系波函数是:
Φ1(1,2)=φA(1)φB(2)实际上两个电子是不可区分的。同样合适的函数是:
Φ2(1,2)=φB(1)φA(2)两个函数Φ1和Φ2都对应相同的能量。海特勒和伦敦就取两个函数的等权线性组合作为H2的变分函数:
Ψ(1,2)=c1Φ1+c2Φ2解久期方程得c1=±c2,波函数和能量是:

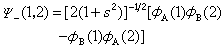
 式中
式中 

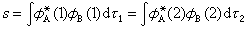 s称原子轨道的重叠积分。算出能量公式中各项,积分得:
s称原子轨道的重叠积分。算出能量公式中各项,积分得: 式中Q、J、s都是R的函数。若用ΔE±表示分子能量与两个分离原子能量之差(图2):
式中Q、J、s都是R的函数。若用ΔE±表示分子能量与两个分离原子能量之差(图2):
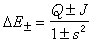 ΔE±就是分子相对于分离原子能量为零时的能量。因为H11和H12都是负量,Ψ+态比Ψ-态能量更低,图2 中ΔE+曲线总处于ΔE-曲线的下面。图中虚线表示实验势能曲线。 ΔE+曲线有极小值,表示形成了稳定的 H2。在平衡核间距 Re=0.87埃,计算得到离解能De=3.14电子伏(或称结合能)。与实验值Re=0.742埃,De=4.75电子伏略有差异,这反映了海特勒-伦敦法的近似程度。ΔE-在R 减小时一直升高。Ψ+称海特勒-伦敦函数,描述H2基态,Ψ-描述排斥态。
ΔE±就是分子相对于分离原子能量为零时的能量。因为H11和H12都是负量,Ψ+态比Ψ-态能量更低,图2 中ΔE+曲线总处于ΔE-曲线的下面。图中虚线表示实验势能曲线。 ΔE+曲线有极小值,表示形成了稳定的 H2。在平衡核间距 Re=0.87埃,计算得到离解能De=3.14电子伏(或称结合能)。与实验值Re=0.742埃,De=4.75电子伏略有差异,这反映了海特勒-伦敦法的近似程度。ΔE-在R 减小时一直升高。Ψ+称海特勒-伦敦函数,描述H2基态,Ψ-描述排斥态。若考虑自旋,按照泡利原理,必须使分子波函数对电子交换是反对称的。则Ψ+必须乘以反对称自旋函数而给出自旋单重态:
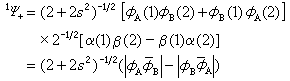 Ψ-必须与对称自旋函数相乘得到自旋三重态:
Ψ-必须与对称自旋函数相乘得到自旋三重态: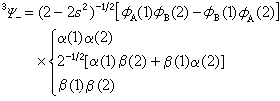 1Ψ+态描述了H2的共价键,其中电子自旋是配对的,故称共价键为电子对键。
1Ψ+态描述了H2的共价键,其中电子自旋是配对的,故称共价键为电子对键。电子密度分布 可以帮助理解共价键的本质。从波函数 Ψ±出发可以计算总电子密度为两个单电子几率密度P±(1)和P±(2)的和乘以电子电量(a,u)。点(x,y,z)处的总电子密度为:

=
 (1)若φA、φB为氢原子的1s轨道,则:
(1)若φA、φB为氢原子的1s轨道,则: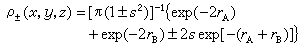 式中rA、rB分别表示从点(x,y,z)到核A和B的距离。总电子电荷密度沿核间轴分布如图3。
式中rA、rB分别表示从点(x,y,z)到核A和B的距离。总电子电荷密度沿核间轴分布如图3。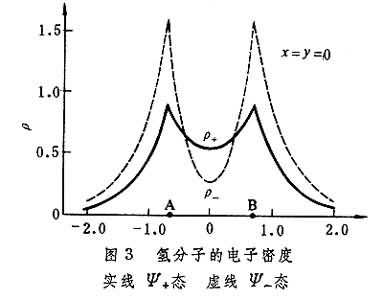 由ρ+曲线可见,电子电荷从核外区移向两核之间的区域,相当于电子同时吸引两核,因而降低了势能。由式(1)可知,两原子核愈接近,重叠积分愈大,电荷在核间区愈密集,也即共价键愈牢固(最大重叠原则)。但原子核愈接近,核排斥能和电子排斥能也同时增加,所以氢分子有一稳定的平衡核间距。Ψ-态的电子电荷从核间区移向核外区,使得核间屏蔽减少,能量升高,形成排斥态。
由ρ+曲线可见,电子电荷从核外区移向两核之间的区域,相当于电子同时吸引两核,因而降低了势能。由式(1)可知,两原子核愈接近,重叠积分愈大,电荷在核间区愈密集,也即共价键愈牢固(最大重叠原则)。但原子核愈接近,核排斥能和电子排斥能也同时增加,所以氢分子有一稳定的平衡核间距。Ψ-态的电子电荷从核间区移向核外区,使得核间屏蔽减少,能量升高,形成排斥态。电子电荷在两核间密集,影响分子的平均动能〈T〉和平均势能〈V〉。为深入理解共价键的本质,按双原子分子的维里定理计算出2的〈T〉和〈V〉:
 又分子总能量E=〈T〉+〈V〉。如已知E 随R 的改变的(dE/dR),则得:
又分子总能量E=〈T〉+〈V〉。如已知E 随R 的改变的(dE/dR),则得:<T>=-[E+R(dE/dR)]
<V>=2E+R(dE/dR)计算得到的H2基态E、<T>、〈V〉都是R 的函数(图4)。

当核间距减少时,电子同核吸引的平均势能降低,但电子的排斥能的平均值增加,核的排斥能也增加。核间距达到某一值(1.401a0, a0为玻尔半径)时,平均总势能达到极小值,电子将在此势阱中运动,此时,dE/dR=0,平均动能等于平均总势能的负值的一半,氢分子的总能量则为势能平均值的一半。
发展 量子力学对电子对键(或共价键)给出了定量的描述。当然,海特勒-伦敦模型只是最早的一个化学键近似处理。从那以后,已经引入许多相当复杂的近似波函数进行较精确的计算,最精确的计算是在1960~1968年由W.科洛斯和C.C.J.罗特汉完成的,他们使用了一个含有100项的函数(考虑了核运动效应),得到离解能计算值De=4.74759电子伏,与实验值De=4.74759电子伏,获得高度的一致。
电子配对法是海特勒-伦敦处理氢分子方法的推广,要点如下:
① 若两原子轨道φA和φB互相重叠,两个轨道上各有一个电子,且电子自旋方向相反,则电子配对给出单重态,形成一个电子对键。
② 两个电子相互配对后,不能再与第三个电子配对,这就是共价键的饱和性。
③ 遵循最大重叠原则,共价键沿着原子轨道重叠最大的方向成键。共价键具有方向性。原子轨道通常在某个特定方向上有最大值,只有在此方向上轨道间才有最大重叠而形成共价键。不同原子轨道有不同的成键能力。在原子轨道φ有最大值的方向上,φ的最大值决定着重叠的大小,因此φ的最大值可做为原子轨道成键能力的度量。鲍林给出s、p、d、f等原子轨道成键能力依次为1、呑、厾、彉。在主量子数相同时,成键能力大的轨道形成的共价键较牢固。
由于异核双原子分子的键有极性,在分子波函数中必须包括离子结构的贡献。如果异核双原子分子AB只有一个电子对键,则有两种可能的离子结构A+B-和A-B+。分子波函数应为:
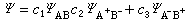 实际问题中只有一种离子结构贡献大些,例如氯化氢HCl分子,由H的1S电子与Cl的3pz电子配对形成共价键,描述它的共价结构的函数是:
实际问题中只有一种离子结构贡献大些,例如氯化氢HCl分子,由H的1S电子与Cl的3pz电子配对形成共价键,描述它的共价结构的函数是: 离子波函数只有:
离子波函数只有: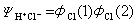 而离子结构
而离子结构 贡献很小,可以略而不计。则HCl的波函数为:
贡献很小,可以略而不计。则HCl的波函数为: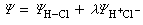
价键理论与化学家所熟悉的经典电子对键概念相吻合,一出现就得到迅速发展。但价键理论计算比较复杂,使得相当长时间内进展缓慢。50年代中国化学家唐敖庆等建议的双电子键函数法也是一项重要进展。60年代,由于发展出直接用基函数计算重叠矩阵和哈密顿矩阵元,不须再使用参数,使得价键理论计算能够程序化。随着计算技术的日益提高,价键理论还将取得新发展。